Abstracts
Résumé
Les méthodes modernes d’analyse du génome ont permis d’aborder des questions posées de longue date telles que les bases moléculaires des anomalies chromosomiques de structure ou la diathèse (prédisposition) aux aneuploïdies. L’architecture du génome révélée par le séquençage permet d’expliquer la récurrence de microremaniements par échange homologue non-allélique entre des répétitions segmentaires formées au cours de l’évolution des primates. Cette particularité structurale du génome a conduit à l’individualisation d’une nouvelle classe de maladies génétiques appelées désordres génomiques par opposition aux maladies géniques dues à des mutations intragéniques. L’étude de l’origine parentale et cellulaire des aneuploïdies éclaire d’un jour nouveau les mécanismes de contrôle de la méiose différents chez l’homme et chez la femme, ainsi que le rôle majeur de l’âge maternel et de la recombinaison pour la répartition méiotique correcte des chromosomes. Ces nouvelles données apportent des clés essentielles pour la compréhension des pathologies chromosomiques chez l’homme.
Summary
Novel methods allowing to analyze the human genome make it possible to assess old questions such as the molecular basis of structural chromosome anomalies and the diathesis to aneuploidy. The architecture of the human genome as unravelled by the human genome sequencing project allows to explain the recurrence of microdeletions and microduplications caused by a non allelic homologous recombination involving segmental duplications created during the evolution of primates. This structural feature of the human genome is associated with a novel class of genetic diseases called genomic disorders as opposed to genetic diseases due to gene mutations. The study of the parental and cellular origin of aneuploidy shed new light on the different mechanisms controlling meiosis in man and woman. In addition it contributes to define the role of maternal age and genetic recombination on the behavior of chromosomes during meiosis. These new data greatly contribute to our understanding of human chromosomal diseases.
Article body
Deux des évolutions marquantes de ces dernières années en cytogénétique ont été l’identification des bases moléculaires de certains remaniements de structure récurrents [1, 2] et celle des mécanismes à l’origine des aneuploïdies[1]. Ces évolutions ont conduit d’une part à l’individualisation d’une nouvelle classe de maladies génétiques, les désordres génomiques, qui regroupent des maladies mendéliennes, des syndromes de gènes contigus et d’autres types de remaniements chromosomiques [3, 4] (Tableau I) et d’autre part, à une meilleure compréhension de l’origine et de la formation des aneuploïdies [5] qui constituent avec les désordres génomiques la première cause connue de handicap mental et physique.
Tableau I
Exemples de désordres génomiques (d’après [4]).
![Exemples de désordres génomiques (d’après [4]).](/en/journals/ms/2005-v21-n11-ms1020/011959ar/media/011959art001n.png)
Répétitions segmentaires, duplicons ou low copy repeats (LCR)
Syndromes de gènes contigus
Les premiers syndromes par microdélétion/duplication ou syndromes de gènes contigus [6] ont été décrits dès 1978 grâce aux techniques de cytogénétique de haute résolution. Ces remaniements de petite taille, inférieure à 5 Mb, le plus souvent, sont associés de façon spécifique à des syndromes cliniques décrits indépendamment de l’anomalie chromosomique. Le phénotype anormal résulte d’un dosage inapproprié de certains gènes dans une région critique. Ce déséquilibre peut résulter d’un mécanisme structurel (délétion ou duplication) ou fonctionnel (empreinte ou disomie uniparentale). Les exemples les plus connus des syndromes microdélétionnels incluent le syndrome de Prader-Willi (PWS) en 15q11-q13, le syndrome de Williams en 7q11, le syndrome de Smith-Magenis (SMS) en 17p11.2, ou le syndrome de DiGeorge/vélocardiofacial (DG/VCFS) en 22q11.2. La découverte de ces déséquilibres spécifiques a ouvert un champ nouveau de la pathologie chromosomique entraînant le développement de nouvelles techniques diagnostiques fondées sur l’hybridation in situ révélée par fluorescence (FISH) et ouvrant de nouvelles voies de recherche. Ces microdélétions/duplications surviennent habituellement de novo avec un taux de mutation élevé dans la population générale. Cette observation suggérait fortement l’existence de facteurs favorisant leur survenue. Le clonage des points de cassure de certains de ces syndromes microdélétionnels a mis en évidence des répétitions d’un type particulier que l’on a appelé CMT1A-REP et SMS-REP sur le chromosome 17, BP1-4 sur le 15, LCR sur le 22…[7]. Ces éléments très particuliers, appelés répétitions segmentaires, duplicons ou encore LCR (low-copy repeats) représentent au moins 5 % de notre génome et constituent la base moléculaire de ces remaniements intrachromosomiques récurrents.
Caractéristiques des répétitions segmentaires
Ce sont des blocs d’ADN de 1 à 400 kb de longueur, formés de segments génomiques présents en plus d’une copie dans le génome et qui ont un degré remarquable d’identité de séquence (95-99 %). Ces blocs peuvent être intrachromosomiques, spécifiques d’un chromosome, ou interchromosomiques situés sur des chromosomes différents. Certaines régions du génome, plus particulièrement les régions péricentromériques et subtélomériques des chromosomes, sont enrichies en ce type de séquences [8].
Structure des répétitions segmentaires
À la différence des autres séquences répétées du génome humain, ces segments reproduisent la structure génomique complète ou partielle de gènes connus, ce qui suggère une transposition récente à partir d’une autre région du génome. Ils ont l’aspect d’un ADN normal et, à première vue, ne sont pas facilement identifiables en tant que séquences répétées. Ces grands segments génomiques sont dits paralogues car ils dérivent d’une duplication au sein d’une même espèce, alors que des gènes dérivant d’un même gène ancestral observés dans des espèces différentes sont dits orthologues. La structure des duplicons peut être assez simple ou complexe, composée de différents modules dupliqués/transposés à partir de chromosomes différents, puis à nouveau dupliqués et remaniés [9] (Figure 1). Par hybridation in situ sur différents génomes et comparaison de séquences, il a été montré que ces blocs se sont formés par transposition duplicative tout au long de l’évolution des primates, c’est-à-dire au cours des dernières 40 millions d’années [10].
Figure 1
Mécanisme en deux étapes proposé pour expliquer la formation de répétitions segmentaires.

Dans un premier temps des segments génomiques dupliqués à partir de différents chromosomes s’accumulent au niveau d’un site accepteur où ils forment une mosaïque de séquences dupliquées. Plus tard, au cours de l’évolution des primates, ces blocs complexes sont eux-mêmes dupliqués et transposés au niveau d’autres régions où ils seront retrouvés sous une forme plus ou moins remaniée (d’après [9]).
Remaniements chromosomiques liés aux duplicons
La forte homologie de séquence des répétitions segmentaires en font un substrat moléculaire de recombinaison homologue non-allélique (NAHR). Cette recombinaison anormale entre des segments répétés spécifiques d’une région ou d’un chromosome entraîne la perte ou la duplication du segment génomique compris entre les deux duplicons et explique la récurrence de certaines pathologies. Différents facteurs incluant la taille des répétitions, l’intervalle qui les séparent, leur degré d’homologie et leur orientation relative influencent la probabilité de misappariement et le type de remaniement généré. D’une façon générale, plus les segments sont grands et plus leur degré d’homologie est élevé, plus la probabilité de survenue d’un échange anormal est grande. Par exemple, le syndrome microdélétionnel le plus fréquent (1/4 000 naissances), la délétion 22q11 responsable du syndrome de DiGeorge/VCF, est engendré par des blocs de répétitions de taille supérieure à 300 kb et dont l’identité de séquence atteint 99,7 % [11].
Une NAHR entre deux répétitions segmentaires intrachromosomiques de même orientation va entraîner une délétion ou une duplication du segment intermédiaire avec une fréquence théoriquement égale (Figure 2A). En fait, on détecte beaucoup plus de délétions que de duplications, peut-être du fait d’un retentissement moindre sur le phénotype. Si les séquences sont en orientation opposée, une NAHR peut entraîner une inversion du segment intermédiaire (Figure 2B). Ces inversions sans conséquence phénotypique sont effectivement retrouvées dans la population générale et chez les parents des enfants porteurs de déséquilibres de la même région [12]. La formation de petits chromosomes métacentriques surnuméraires dérivés du 15 et du 22 correspond également à des NAHR entre des séquences en orientation inverse au niveau de duplicons (Figure 2C).
Figure 2

A. Un échange entre des séquences homologues non-alléliques en orientation directe, distantes de quelques kilobases à plusieurs mégabases, entraîne une délétion ou une duplication du segment intermédiaire. B. Un échange entre des séquences homologues non-alléliques en orientation inverse sur une même chromatide entraîne la formation d’une inversion paracentrique. C. Un échange entre des séquences en orientation inverse entre deux chromatides entraîne la formation d’un chromosome dicentrique. Si les chromosomes impliqués sont des acrocentriques, l’élément formé sera porteur d’un satellite à chaque extrémité.
Remaniements interchromosomiques et non-récurrents
Des études plus récentes ont montré que les répétitions segmentaires ou d’autres éléments de l’architecture du génome peuvent aussi être à l’origine de remaniements interchromosomiques récurrents [13, 14]. Un certain nombre de remaniements non-récurrents ont aussi comme base moléculaire des duplicons [15].
Aneuploïdies
Origine parentale et cellulaire des aneuploïdies
Au cours des dernières années, les polymorphismes de l’ADN ont été utilisés pour déterminer l’origine parentale et cellulaire des différentes aneuploïdies [16].
Pour les monosomies, une information n’est disponible que pour la monosomie X qui est en pratique la seule monosomie observée à la naissance. Plusieurs études sur l’origine de la monosomie X ont montré que, dans 80 % des cas, le chromosome X conservé était d’origine maternelle [17]. Par voie de conséquence, le chromosome Y ou le chromosome X d’origine paternelle est perdu au cours de la méiose ou plus vraisemblablement dans les premières divisions du zygote. Les résultats des études menées sur les fausses couches sont identiques. Cela suggère qu’il n’existe pas de différence dans la survie de l’embryon/foetus monosomique en fonction de l’origine parentale du chromosome X [18].
L’origine de la trisomie 21 a été la plus étudiée (Tableau II). Le résultat de ces études montre que 90 % des trisomies 21 résultent d’une erreur au cours de la méiose maternelle. Environ 10 % des cas résultent d’une erreur paternelle et dans 2 % des cas il existe une nondisjonction mitotique postzygotique [19]. En outre, la fréquence et peut-être le mécanisme de nondisjonction varient en fonction du contexte gamétique puisque dans l’ovocyte, une erreur de première division méiotique (MI) est trois fois plus fréquente qu’une erreur de deuxième division méiotique (MII), tandis qu’elles sont de fréquence égale dans les spermatocytes [20].
Tableau II
Origine des trisomies.
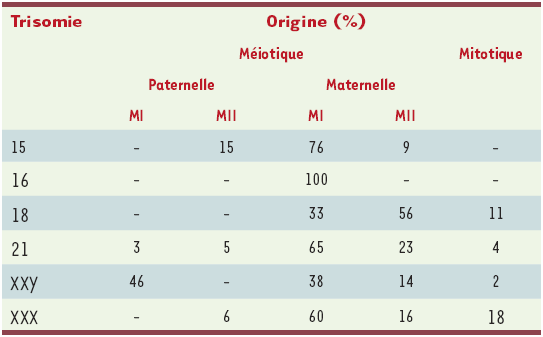
MI : méiose I ; MII : méiose II (adapté de [5]).
L’origine parentale et méiotique des autres aneuploïdies a fait également l’objet de plusieurs investigations (pour revue voir [5]). Deux grands principes semblent émerger. Premièrement, il existe une grande variation d’une trisomie à l’autre concernant l’origine parentale du chromosome additionnel. Par exemple, les erreurs d’origine paternelle touchent 50 % des 47, XXY, mais seulement 5-10 % des autres trisomies. Elles sont pratiquement absentes dans les trisomies 16. Deuxièmement, la proportion d’erreurs de MI ou de MII varie d’un chromosome à l’autre. Par exemple, parmi les trisomies d’origine maternelle, la totalité des trisomies 16 et un tiers des polysomies X résultent d’une erreur de première division méiotique, tandis que 50 % des trisomies 18 résultent d’une erreur de deuxième division méiotique.
Mécanismes méiotiques
Warren a été le premier à montrer qu’une diminution de la recombinaison génétique était associée à la nondisjonction [21]. Depuis, un grand nombre de laboratoires ont confirmé cette observation. Les études les plus détaillées concernent les trisomies 15, 16, 18 et 21 d’origine maternelle, ainsi que celles impliquant les chromosomes sexuels et les trisomies 21 ou le génotype XXY d’origine paternelle. Cette diminution de recombinaison varie considérablement d’un chromosome à l’autre. Par exemple, la réduction est très prononcée dans le syndrome XXY d’origine paternelle. En effet, dans ce cas, la carte génétique de la région pseudo-autosomique est réduite de 50cM à 10cM [22]. Pour d’autres trisomies (comme la trisomie 15), l’effet est moindre [23].
Deux phénomènes peuvent être responsables de cette diminution de recombinaison. Tout d’abord, il peut exister des situations où aucune recombinaison n’est observée. Ensuite, la nature de la diminution peut être complexe. Ainsi, pour un bivalent présentant habituellement trois chiasmata, outre une réduction du nombre des chiasmata, la disposition de ceux-ci sur le bivalent peut être atypique. En fait, les deux phénomènes sont observés bien que leurs contributions respectives varient en fonction du chromosome impliqué. Par exemple, une absence d’échange (ou une absence d’appariement) est observée dans environ 40 % des cas de trisomie 21 résultant d’une erreur maternelle en première division méiotique. Cela est également observé dans le cas de la trisomie 21 et du syndrome XXY d’origine paternelle ainsi que dans le cas des trisomies 21, 15, 18 ou les polysomies X d’origine maternelle. Mais dans les autres cas de trisomie 21 résultant d’une erreur maternelle de première division méiotique, il existe une disposition anormale des échanges sur le bivalent [19]. Par exemple, lorsqu’un seul échange est présent, cet échange est habituellement positionné de façon télomérique rendant le bivalent très instable sur la plaque équatoriale. Cela est également observé dans les trisomies 16. Enfin, outre une absence de recombinaison ou une disposition inhabituelle des échanges, les études concernant la trisomie 21 montrent également un excès de recombinaison péricentromérique dans les non-disjonctions classées comme des erreurs résultant d’une erreur de deuxième division méiotique. Il est possible que cet excès de recombinaison péricentromérique produise un enchevêtrement chromosomique entraînant une non-disjonction de première division méiotique de l’ensemble du bivalent [24]. Celui-ci se sépare ensuite de manière équationnelle mimant ainsi une non-disjonction de deuxième division méiotique alors que l’erreur méiotique s’est en réalité produite au cours de la première division méiotique (Figure 3). Il a aussi été proposé que ces « échanges » péricentromériques puissent interférer avec la cohésion des chromatides-soeurs causant une division prématurée des chromatides en première division méiotique. Si ces chromatides migrent ensuite vers le même pôle cellulaire en première et deuxième division méiotique, un gamète disomique résultant apparemment d’une erreur de deuxième division méiotique sera produit. Bien que cela soit le cas pour la trisomie 21, des études plus récentes montrent que le chromosome 18 et les chromosomes sexuels se comportent différemment. En somme, toutes ces études montrent bien que la relation entre anomalie de recombinaison et non-disjonction est complexe.
Figure 3

A.Méiose normale. B, C, D. Différents mécanismes de formation des aneuploïdies lors de la méiose.
Alors que plusieurs laboratoires faisaient appel aux techniques de biologie moléculaire, d’autres investigateurs ont utilisé les techniques de cytogénétique classique ou moléculaire pour analyser directement la ségrégation méiotique des chromosomes. Ces études portaient sur les ovocytes et avaient pour but de déterminer si les erreurs de première division méiotique résultaient d’une non-disjonction ou d’une division prématurée des chromatides-soeurs [25]. Les études les plus récentes montrent que ce deuxième mode de ségrégation des chromosomes, d’abord contesté, est bien réel, mais il n’explique qu’environ un tiers des erreurs méiotiques. En outre, cette proportion varie en fonction de l’âge et des chromosomes impliqués [26, 27].
Cause des aneuploïdies
Après de nombreuses études, l’âge maternel reste le seul facteur causal dont la démonstration ait été faite dans la trisomie et l’aneuploïdie. Cette relation a été décrite par Penrose il y a plus de 60 ans, bien avant que la base chromosomique du syndrome de Down ne soit élucidée [28]. L’augmentation de la fréquence de la trisomie 21 à la naissance est d’abord modérée, passant de 0,05 % à l’âge de 20 ans jusqu’à 0,1 % à 30 ans. Elle est ensuite beaucoup plus rapide, passant de 0,25 % à 35 ans jusqu’à 3 % à 45 ans, suggérant qu’il existe deux composantes à cet effet, l’une qui est indépendante de l’âge maternel et l’autre qui en est dépendante. Par la suite, l’étude des autres trisomies observées dans l’espèce humaine a montré que la grande majorité d’entre elles étaient également influencées par l’âge maternel.
Dans l’ensemble, l’importance de cet effet de l’age maternel est considérable puisqu’à 25 ans, environ 2 % des grossesses sont trisomiques, mais à 40 ans deux grossesses sur trois sont aneuploïdes [29]. En outre, l’effet est spécifique du chromosome impliqué. Par exemple, l’effet de l’âge maternel est important pour les trisomies impliquant les plus petits chromosomes mais est absent pour les plus grands. Enfin la trisomie 16 se comporte de façon particulière dans cette relation puisque sa fréquence augmente de façon linéaire avec l’âge maternel (la composante indépendante de l’âge maternel est apparemment absente) et non pas de façon exponentielle comme c’est le cas pour la plupart des autres trisomies. Les études ont également porté sur les cas de trisomie en mosaïque et ceux présentant une double trisomie. En effet, l’incidence de ces trisomies est aussi affectée par une augmentation de l’âge maternel, l’effet étant le plus important pour les cas de double trisomie [30].
En dépit de ces avancées, la base biologique de cet effet de l’âge maternel reste largement incomprise. Les études de l’origine parentale des aneuploïdies montrent que cet effet est lié aux erreurs d’origine maternelle et non à celles d’origine paternelle. Il semble donc clair que l’ovaire et non pas l’utérus soit la source de cet effet de l’âge maternel.
En outre, les études semblent indiquer que la première division méiotique est particulièrement vulnérable. Un modèle récent propose donc l’existence de deux évènements. Le premier est indépendant de l’âge maternel puisqu’il survient dans l’ovaire foetal et implique la formation d’un bivalent instable. Le second évènement est dépendant de l’âge maternel. Il survient dans l’ovaire adulte même lorsque le bivalent est stable [31].
Outre l’âge maternel, un grand nombre de facteurs génétiques ou de l’environnement ont également été incriminés dans la diathèse[2] aux aneuploïdies. Par exemple, une association entre métabolisme des folates et non-disjonction a été proposée [32]. En particulier, l’étude de la fréquence d’un polymorphisme de la MTHFR, une enzyme impliquée dans le métabolisme des folates, a montré un excès d’hétérozygotes et d’homozygotes pour ce polymorphisme chez les mères d’enfants trisomiques. Des résultats semblables ont été obtenus pour une autre enzyme, la MTRR [33]. Enfin, la présence combinée d’un polymorphisme pour ces deux enzymes semble augmenter la force de cette association. Ces études semblent indiquer qu’une méthylation anormale pourrait être un facteur de risque de non-disjonction et qu’un supplément en acide folique pourrait atténuer ce risque de façon significative. Malheureusement des études plus récentes viennent infirmer cette hypothèse [34].
Conclusions
Les développements de la cytogénétique moléculaire, qui vont des techniques maintenant courantes de FISH jusqu’à la CGH (comparative genomic hybridisation)-array, ont fait l’objet de plusieurs revues [35, 36]. Ils sont déterminants pour l’évolution de notre discipline. Mais ces progrès techniques, quelle que soit leur dimension, ne doivent pas faire oublier les objectifs sous-jacents qui sont la compréhension des mécanismes de survenue des anomalies chromosomiques. Cette connaissance reste indispensable dans un objectif de prévention.
Appendices
Notes
-
[1]
Les aneuploidies correspondent à toute déviation du nombre des chromosomes par rapport à un multiple exact de l’état haploïde (23 chromosomes chez l’homme). Elles comprennent les trisomies et les monosomies.
-
[2]
Diathèse : extériorisation phénotypique d’un génotype anormal, catractérisé par une prédisposition à contracter certaines maladies [37].
Références
- 1. Eichler EE. Masquerading repeats : paralogous pitfalls of the human genome. Genome Res 1998 ; 8 : 758-62.
- 2. Ji Y, Eichler EE, Schwartz S, Nicholls RD. Structure of chromosomal duplicons and their role in mediating human genomic disorders. Genome Res 2000 ; 10 : 597-610.
- 3. Lupski JR. Genomic disorders : structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet 1998 ; 14 : 417-22.
- 4. Stankiewicz P, Lupski JR. Genome architecture, rearrangements and genomic disorders. Trends Genet 2002 ; 18 : 74-82.
- 5. Hassold T, Hunt P. To err (meiotically) is human : the genesis of human aneuploidy. Nat Rev Genet 2001 ; 2 : 280-91.
- 6. Schmickel RD. Contiguous gene syndromes : a component of recognizable syndromes. J Pediatr 1986 ; 109 : 231-41.
- 7. Emanuel BS, Shaikh TH. Segmental duplications : an expanding role in genomic instability and disease. Nat Rev Genet 2001 ; 2 : 791-800.
- 8. Bailey JA, Gu Z, Clark RA, et al. Recent segmental duplications in the human genome. Science 2002 ; 297 : 1003-7.
- 9. Horvath JE, Bailey JA, Locke DP, Eichler EE. Lessons from the human genome : transitions between euchromatin and heterochromatin. Hum Mol Genet 2001 ; 10 : 2215-23.
- 10. Samonte RV, Eichler EE. Segmental duplications and the evolution of the primate genome. Nat Rev Genet 2002 ; 3 : 65-72.
- 11. Bailey JA, Yavor AM, Viggiano L, et al. Human-specific duplication and mosaic transcripts : the recent paralogous structure of chromosome 22. Am J Hum Genet 2002 ; 70 : 83-100.
- 12. Giglio S, Broman KW, Matsumoto N, et al. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet 2001 ; 68 : 874-83.
- 13. Edelmann L, Spiteri E, Koren K, et al. AT-rich palindromes mediate the constitutional t(11;22) translocation. Am J Hum Genet 2001 ; 68 : 1-13.
- 14. Giglio S, Calvari V, Gregato G, et al. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16 ; p23) translocation. Am J Hum Genet 2002 ; 71 : 276-85.
- 15. Shaw CJ, Lupski JR. Implications of human genome architecture for rearrangement-based disorders : the genomic basis of disease. Hum Mol Genet 2004 ; 13 : R57-64.
- 16. Chakravarti A, Majumder PP, Slaugenhaupt SA, et al. Gene-centromere mapping and the study of non-disjunction in autosomal trisomies and ovarian teratomas. In : Hassold T, Epstein C, eds. Molecular and cytogenetic studies of non-disjunction. New York : Alan R. Liss, 1989 : 45-79.
- 17. Jacobs P, Dalton P, James R, et al. Turner syndrome : a cytogenetic and molecular study. Ann Hum Genet 1997 ; 61 : 471-83.
- 18. Hassold T, Pettay D, Robinson A, Uchida I. Molecular studies of parental origin and mosaicism in 45, X conceptuses. Hum Genet 1992 ; 89 : 647-52.
- 19. Lamb NE, Freeman SB, Savage-Austin A, et al. Susceptible chiasmate configurations of chromosome 21 predispose to non-disjunction in both maternal meiosis I and meiosis II. Nat Genet 1996 ; 14 : 400-5.
- 20. Savage AR, Petersen MB, Pettay D, et al. Elucidating the mechanisms of paternal non-disjunction of chromosome 21 in humans. Hum Mol Genet 1998 ; 7 : 1221-7.
- 21. Warren AC, Chakravarti A, Wong C, et al. Evidence for reduced recombination on the nondisjoined chromosomes 21 in Down Syndrome. Science 1987 ; 237 : 652-4.
- 22. Hassold TJ, Sherman SL, Pettay D, et al. XY chromosome nondisjunction in man in associated with diminished recombination in the pseudoautosomal region. Am J Hum Genet 1991 ; 49 : 253-60.
- 23. Robinson WP, Bernasconi F, Mutirangura A, et al. Nondisjunction of chromosome 15 : origin and recombination. Am J Hum Genet 1993 ; 53 : 740-51.
- 24. Bridges CB. Nondisjonction as proof of the chromosome theory of heredity. Genetics 1916 ; 1 : 1-52, 107-63.
- 25. Angell R. First-meiotic-division nondisjunction in human oocytes. Am J Hum Genet 1997 ; 61 : 23-32.
- 26. Dailey T, Dale B, Cohen J, Munne S. Association between nondisjunction and maternal age in meiosis-II human oocytes. Am J Hum Genet 1996 ; 59 : 176-84.
- 27. Anahory T, Andreo B, Regnier-Vigouroux G, et al. Sequential multiple probe fluorescence in-situ hybridization analysis of human oocytes and polar bodies by combining centromeric labelling and whole chromosome painting. Mol Hum Reprod 2003 ; 9 : 577-85.
- 28. Penrose L. The relative effects of paternal and maternal age in mongolism. J Genet 1993 ; 27 : 219-24.
- 29. Hassold T, Chiu D. Maternal age-specific rates of numerical chromosome abnormalities with special reference to trisomy. Hum Genet 1985 ; 70 : 11-7.
- 30. Risch N, Stein Z, Kline J, Warburton D. The relationship between maternal age and chromosome size in autosomal trisomy. Am J Hum Genet 1986 ; 39 : 68-78.
- 31. Lamb NE, Yu K, Shaffer J, et al. Association between maternal age and meiotic recombination for trisomy 21. Am J Hum Genet 2005 ; 76 : 91-9.
- 32. James SJ, Pogribna M, Pogribny IP, et al. Abnormal folate metabolism and mutation in the methylenetetrahydrofolate reductase gene may be maternal risk factors for Down syndrome. Am J Clin Nutr 1999 ; 70 : 495-501.
- 33. Hobbs CA, Sherman SL, Yi P, et al. Polymorphisms in genes involved in folate metabolism as maternal risk factors for Down syndrome. Am J Hum Genet 2000 ; 67 : 623-30.
- 34. Chadefaux-Vekemans B, Coudé M, Muller F, et al. Methylenetetrahydrofolate reductase polymorphism in the etiology of Down syndrome. Pediatr Res 2002 ; 51 : 766-7.
- 35. Romana SP, Gosset P, Elghezal H, et al. Apport de la cytogénétique moléculaire au diagnostic pré et postnatal des anomalies chromosomiques. J Gynecol Obstet Biol Reprod (Paris) 2001 ; 30 (suppl 1) : 75-9.
- 36. Sanlaville D, Lapierre JM, Turleau C, et al. Molecular karyotyping in human constitutional cytogenetics. Eur J Med Genet 2005 (sous presse).
- 37. Garrod AE. The inborn factors in disease. Oxford : Clarendon Press : 1931.
List of figures
Figure 1
Mécanisme en deux étapes proposé pour expliquer la formation de répétitions segmentaires.
Dans un premier temps des segments génomiques dupliqués à partir de différents chromosomes s’accumulent au niveau d’un site accepteur où ils forment une mosaïque de séquences dupliquées. Plus tard, au cours de l’évolution des primates, ces blocs complexes sont eux-mêmes dupliqués et transposés au niveau d’autres régions où ils seront retrouvés sous une forme plus ou moins remaniée (d’après [9]).
Figure 2
A. Un échange entre des séquences homologues non-alléliques en orientation directe, distantes de quelques kilobases à plusieurs mégabases, entraîne une délétion ou une duplication du segment intermédiaire. B. Un échange entre des séquences homologues non-alléliques en orientation inverse sur une même chromatide entraîne la formation d’une inversion paracentrique. C. Un échange entre des séquences en orientation inverse entre deux chromatides entraîne la formation d’un chromosome dicentrique. Si les chromosomes impliqués sont des acrocentriques, l’élément formé sera porteur d’un satellite à chaque extrémité.
Figure 3
A.Méiose normale. B, C, D. Différents mécanismes de formation des aneuploïdies lors de la méiose.
List of tables
Tableau I
Exemples de désordres génomiques (d’après [4]).
Tableau II
Origine des trisomies.
MI : méiose I ; MII : méiose II (adapté de [5]).