
Corps de l’article
La kallicréine tissulaire est la principale enzyme impliquée dans la production des kinines par clivage enzymatique du kininogène. Le système kininogène-kallikréine-kinine est présent dans les cellules endothéliales et musculaires lisses de la paroi vasculaire où les kinines générées localement ont un puissant effet vasodilatateur dépendant de l'endothélium (Figure 1). Cet effet est lié à l'activation du récepteur B2 de la bradykinine, couplé à la libération de monoxyde d'azote (NO), de prostacycline et de facteurs hyperpolarisants. L'inactivation du gène codant pour la kallicréine chez la souris a montré l'importance du système kallicréine-kinine dans la physiologie artérielle : les souris déficientes en kallicréine ont, en effet, un dysfonctionnement endothélial avec une perte de la vasodilatation liée au flux, un processus important de régulation de l'apport sanguin aux organes [1, 2]. La kallicréine est également synthétisée en abondance dans le rein, dans la partie terminale du tubule distal et la partie corticale du tubule collecteur. Elle est libérée dans l'urine et dans la circulation et l'interstitium péritubulaire. Le système kallicréine-kinine rénal agit de concert avec le système rénine-angiotensine pour réguler le flux sanguin médullaire et papillaire. L'excrétion urinaire de la kallicréine reflète la synthèse de l'enzyme par le rein. On sait depuis de nombreuses années que l'activité kallicréine urinaire est influencée par des facteurs génétiques, car il existe une ressemblance familiale de cette activité. Elle est régulée également par les apports sodiques et potassiques.
Figure 1
Le système kallicréine-kinine.
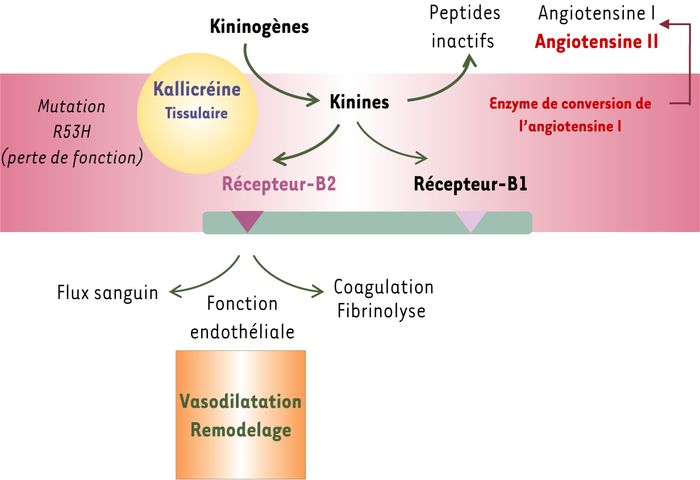
Le système kininogène-kallikréine-kinine est présent dans les cellules endothéliales et musculaires lisses de la paroi vasculaire. Les kinines, produites localement à partir des kininogènes, activent le récepteur B2 de la bradykinine. Cette activation induit un puissant effet vasodilatateur qui est dépendant de l'endothélium.
L'étude moléculaire du gène humain codant pour la kallicréine (hKLK1) a permis récemment de mettre en évidence plusieurs polymorphismes siégeant sur les parties codantes et non-codantes du gène. Parmi les différentes variations observées, la mutation faux-sens R53H est associée à une réduction de l'ordre de 50 % de l'activité urinaire de kallicréine chez les sujets hétérozygotes. La synthèse d'un variant recombinant de la kallicréine porteur de cette mutation montre qu'elle entraîne un effondrement de l'activité enzymatique réduite à 1 % de celle de la protéine sauvage. Cela s'explique par la situation de l'Arg 53 dans un sous-site de liaison du substrat [3]. Il s'agit de la première mutation fonctionnelle identifiée dans le système kallicréine-kinine chez l'homme, et l'une des bases moléculaires de la ressemblance familiale de l'activité kallicréine urinaire décrite auparavant. Cette mutation est présente, à l'état hétérozygote, chez 5 à 7 % des sujets appartenant aux populations d'origine caucasienne, et 14 % de ceux appartenant aux populations d'origine africaine.
L'objectif de l'étude clinique [4] réalisée chez les sujets porteurs de la mutation R53H, qui constituent donc un « modèle » humain de déficience en kallicréine, est de confirmer chez l'homme le rôle de cette enzyme dans la fonction artérielle, à la suite des observations faites chez la souris, et de décrire les conséquences vasculaires, rénales et hormonales d'un déficit congénital partiel en activité kallicréine. L'exploration de la fonction endothéliale a été réalisée de façon non invasive au niveau de l'artère brachiale, avec étude de la vasodilatation dépendante du flux survenant après levée de l'ischémie distale au poignet induite par un brassard gonflé pendant cinq minutes au-dessus de la pression systolique, et l'étude de la réponse indépendante à l'endothélium engendrée par la trinitrine sublinguale. Les mesures ont été réalisées dans deux conditions contrastées d'apport de sodium et de potassium (régimes riche en sodium et pauvre en potassium, et pauvre en sodium et riche en potassium) pour moduler la synthèse de kallicréine.
Les résultats montrent que la réponse vasodilatatrice endothélium-dépendante au flux ainsi que la réponse à la trinitrine, ne sont pas modifiés chez les sujets R53H hétérozygotes par rapport aux sujets homozygotes R53R. En revanche, les sujets hétérozygotes présentent une augmentation permanente des forces de cisaillements (shear stress), associée avec une réduction paradoxale (car on attendrait, en réponse à cette anomalie une expansion luminale) du diamètre de l'artère brachiale, témoignant d'un dysfonctionnement endothélial chronique. Ces résultats confirment l'importance de la kallicréine dans la fonction artérielle chez l'homme, et mettent en évidence une nouvelle forme de dysfonctionnement artériel, affectant 5 à 7 % de la population. Les sujets étudiés étaient des hommes jeunes et normotendus. Ces anomalies artérielles pourraient cependant prédisposer à long terme au développement des maladies vasculaires dégénératives. En outre, les souris déficientes en kallicréine ont perdu, en raison de la disparition des kinines, une partie des mécanismes de cardioprotection mis en oeuvre lors de l'ischémie myocardique expérimentale [5]. Les sujets R53H pourraient donc avoir un risque accru de cardiopathie ischémique et d'infarctus du myocarde, hypothèse à confirmer par des études cliniques complémentaires. Sur le plan rénal, les sujets R53H s'adaptent normalement aux variations en sodium et en potassium des régimes. Ils ne présentent pas d'anomalies de la régulation du système rénine-angiotensine-aldostérone, ou des facteurs natriurétiques auriculaires. Ils présentent donc un déséquilibre permanent dans le rein entre l'activité du système rénine-angiotensine et celle du système kallicréine-kinine, dont les conséquences possibles à long terme restent à déterminer.
Parties annexes
Références
- 1. Meneton P, Bloch-Faure M, Hagege AA, et al. Cardiovascular abnormalities with normal blood pressure in tissue kallikrein-deficient mice. Proc Natl Acad Sci USA 2001 ; 98 : 2634-9.
- 2. Bergaya S, Meneton P, Bloch-Faure M, et al. Decreased flow-dependent dilation in carotid arteries of tissue kallikrein-knockout mice. Circ Res 2001 ; 88 : 593-9.
- 3. Slim R, Torremocha F, Moreau T, et al. Loss-of-function polymorphism of the human kallikrein gene with reduced urinary kallikrein activity. J Am Soc Nephrol 2002 ; 13 : 968-76.
- 4. Azizi M, Boutouyrie P, Bissery A, et al. Arterial and renal consequences of partial genetic deficiency in tissue kallikrein activity in humans. J Clin Invest 2005 ; 115 : 780-7.
- 5. Griol-Charbidili V, Messadi-Laribi E, Bascands JL, et al. Role of tissue kallikrein in the cardioprotective effects of ischemic and pharmacological preconditioning in myocardial ischemia. FASEB J 2005 online.
Liste des figures
Figure 1
Le système kallicréine-kinine.
Le système kininogène-kallikréine-kinine est présent dans les cellules endothéliales et musculaires lisses de la paroi vasculaire. Les kinines, produites localement à partir des kininogènes, activent le récepteur B2 de la bradykinine. Cette activation induit un puissant effet vasodilatateur qui est dépendant de l'endothélium.