Résumés
Résumé
L’obésité, en particulier ses formes sévères, est l’un des principaux facteurs de risque de diabète de type 2. Par ailleurs, l’explosion de l’épidémie d’obésité infantile s’accompagne de l’apparition de formes précoces de diabète de type 2, avec un syndrome métabolique et une intolérance au glucose détectables dès l’adolescence. Ces données suggèrent l’existence de déterminants moléculaires primitifs communs entre obésité sévère et précoce et diabète de type 2, qui partageraient une insulinorésistance génétiquement déterminée. Dans ce contexte, l’identification, sur la région chromosomique 6q liée à la « diabésité », du gène ENPP1 codant pour un inhibiteur du récepteur de l’insuline, dont des variants codants et non codants contribuent au risque génétique de cette affection, est un pas vers la dissection génétique des obésités les plus diabétogènes. Ces résultats ouvrent des perspectives nouvelles vers le profilage génétique et biologique des adolescents obèses, dans une optique de prévention et de traitement de la « diabésité » et de ses complications vasculaires.
Summary
Clinical studies have established the strong link between obesity and type 2 diabetes, especially in children, where the rising prevalence of childhood severe obesity has preceded the recent emergence of early-onset forms of “diabesity”. These data suggested a common genetic background shared by both conditions, which was also supported by the identification by genome scans of several diabesity chromosomal regions of linkage. The genetic investigation of early-onset form of familial obesity linkage to chromosome 6q led to the identification of ENPP1, an inhibitor of the insulin receptor, as a possible molecular mechanism behind both obesity and type 2 diabetes. Analysis of the DNA variations of ENPP1 in 6,147 subjects showed association between a combination of variants and both childhood obesity, morbid or moderate obesity in adults and also with type 2 diabetes. This study provides a first molecular basis for the physiopathologic association between severe insulin resistance and obesity, and further type 2 diabetes, and offers a new perspective for prevention and treatment of these conditions.
Corps de l’article
La prévalence de l’obésité a augmenté de façon alarmante ces deux dernières décennies [1], et pourrait être la cause directe d’une diminution générale de 2 à 5 ans de la longévité dans les prochaines années [2]. L’obésité modérée de l’adulte, définie par un indice de masse corporelle (IMC) supérieur ou égal à 30 kg/m2, est passée en France de 8,2 % à 11,3 % entre 1997 et 2003, soit une augmentation de 37,8 % [2]. Dans le même temps, l’obésité massive (IMC ≥ 40 kg/m2) montrait une augmentation de 100 % (0,3 % à 0,6 %) [2]. Si on est loin encore des chiffres américains (30 % d’obèses avec près de 5 % d’obésité massive), la tendance est la même. Si des changements d’ordre environnemental (alimentation déséquilibrée, sédentarité) sont essentiellement en cause dans l’augmentation de la corpulence de la population générale, l’explosion des formes extrêmes d’obésité suggère l’existence d’une prédisposition génétique. Les études familiales confirment d’ailleurs que l’héritabilité (variance génétique rapportée à la variance phénotypique) de l’obésité se situe autour de 80 % [3].
Le risque de diabète de type 2 (DT2) croit avec le degré d’obésité. Ainsi, Mokdad et al., dans une étude portant sur 195 005 Américains, établit que le risque relatif de DT2 est de 3,44 chez les individus obèses modérés, et d’un facteur de 7,37 chez les individus obèses massifs [4]. L’apparition récente de cas de DT2 chez les adolescents et même chez certains enfants est directement corrélée avec l’explosion de la prévalence de l’obésité infantile [5]. Cependant, tous les obèses ne deviennent pas diabétiques. Comme le DT2 a lui-même une base génétique, l’existence de déterminants moléculaires communs entre obésité et DT2 a été postulée depuis longtemps mais jamais vraiment étayée. Ainsi les formes monogéniques de DT2 (diabète MODY) ne sont pas accompagnées d’obésité, et les formes mendéliennes d’obésité avec atteinte de la voie de la leptine-mélanocortine ne sont pas particulièrement diabétogènes. Cependant, les analyses génomes entiers de familles diabétiques ou obèses ont montré quatre régions du génome, situées sur les chromosomes 6, 8, 10 et 20, et présentant des liaisons génétiques redondantes simultanément avec l’obésité et le DT2, suggérant que dans ces régions pourraient se trouver des gènes candidats communs.
Le recrutement de 115 familles françaises avec obésité infantile a d’abord permis de publier la 1re étude génome entier pour des traits associés à l’obésité infantile, qui montrait une liaison génétique sur les chromosomes 2, 6, 15, 16, 17 et 19, le plus fort signal de liaison se situant dans la région 6q22.31-q23.2 [6]. Premier élément frappant, l’analyse des 62 familles participant à la liaison génétique au chromosome 6 a révélé des perturbations de l’homéostasie glucidique chez les enfants obèses et leurs parents comparativement aux autres familles. Les enfants présentaient des glycémies plus élevées durant le test d’hyperglycémie provoquée par voie orale, un indice insulinogénique plus faible, signe d’un trouble de la 1re phase sécrétoire de l’insuline, et 3 % des enfants des 62 familles (versus 0 %) étaient intolérants au glucose ou diabétiques. De plus, 14 % des parents des familles liées au chromosome 6 étaient diabétiques contre 3 % dans les autres familles. Ces données suggéraient que le(s) gène(s) de susceptibilité à l’obésité sur le chromosome 6 pouvaient également influer sur l’homéostasie du glucose. L’intervalle de confiance de la liaison génétique avec l’obésité infantile couvrait 41,4 Mb et incluait 166 gènes référencés. Cependant, la comparaison de cet intervalle avec ceux de sept autres études montrant chez l’adulte une liaison avec des traits associés à l’obésité [7] ou au DT2 [8-13] a permis d’identifier une région de 2,4 Mb commune à toutes ces études, localisée entre les deux marqueurs microsatellites D6S1656 et D6S270, incluant 22 gènes référencés (Figure 1).
Figure 1
Un intervalle de 2,4 Mb commun à huit études génome entier de « diabésité ».

ENPP1, un gène candidat de la diabésité sur le chromosome 6
Le meilleur gène candidat dans l’intervalle de 2,4 Mb était le gène de l’ectonucléotide pyrophosphatase phosphodiestérase 1 (ENPP1). En effet, un des rôles suggérés d’ENPP1 serait d’inhiber le récepteur de l’insuline par interaction directe, affectant du même coup la cascade de signalisation en aval du récepteur [14, 15]. En outre, un polymorphisme fonctionnel de l’exon 4 (K121Q), entraînant une inhibition plus marquée du récepteur de l’insuline [16], a été associé à l’insulinorésistance dans diverses ethnies [17, 18]. Argument supplémentaire en faveur d’un rôle potentiel d’ENPP1 dans la susceptibilité à l’obésité infantile, le marqueur D6S1656, situé dans l’intron 1 d’ENPP1, réplique une liaison génétique dans un 2e groupe indépendant de 68 familles avec obésité infantile. Ce marqueur montre également une association familiale avec l’obésité infantile dans les deux groupes de familles testés.
Les régions codantes et 1 300 paires de bases en amont du codon d’initiation ATG et en aval du codon stop TGA ont été d’abord séquencées chez des enfants obèses des familles liées au chromosome 6 et chez des adultes témoins. Quarante et un polymorphismes ont été identifiés, dont 22 fréquents. L’analyse du déséquilibre de liaison au locus ENPP1 a montré que le génotypage de 10 tagged polymorphismes était suffisant pour extraire l’essentiel de l’information génétique. En outre, 25 fragments de haute homologie (> 70%) entre l’homme, le rat et le poisson Fugu, situés dans les régions introniques et intergéniques d’ENPP1, ont permis d’identifier 11 polymorphismes supplémentaires qui pouvaient avoir un rôle fonctionnel.
Les variants d’ENPP1 sont associés avec l’obésité
Les polymorphismes d’ENPP1 ont ensuite été génotypés chez des enfants obèses, des adultes obèses morbides et des adultes non obèses et normo-tolérants au glucose. Les études cas-témoin ont établi que 6 polymorphismes d’ENPP1 (IVS2 delG + 8, K121Q, IVS8 T > G + 27, IVS20 delT - 11, A > G + 1044 TGA, T > G + 5954 TGA) étaient associés à l’obésité sévère, le résultat le plus significatif étant obtenu pour le K121Q (odd ratio = 1,37, p = 0,00008). Afin de confirmer que l’association avec l’obésité était bien spécifique de ENPP1, 55 polymorphismes dans une région de 580 kb incluant 4 gènes adjacents à ENPP1 ont été étudiés, montrant l’existence de 3 blocs de déséquilibre de liaison distincts (Figure 2). Huit polymorphismes ont été associés simultanément à l’obésité infantile et à l’obésité infantile liée au chromosome 6q, dont 7 étaient inclus dans un bloc limité à ENPP1(Figure 3). Ces résultats confirment que l’association avec l’obésité est bien due aux polymorphismes d’ENPP1.
Figure 2
Carte de déséquilibre de liaison pour 53 SNP répartis sur une région de 580 kilobases incluant les gènes ARG1, CRSP3, ENPP3, ENPP1 et CTGF.
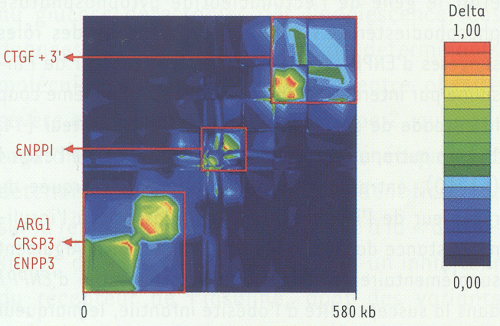
Le génotypage des 53 SNP a été réalisé chez 298 individus témoins et 421 enfants obèses. Les régions de haut et de bas déséquilibre de liaison (indice delta) sont représentées respectivement par les couleurs rouges et bleues. La carte est à l’échelle physique.
Figure 3
Association des 53 SNP répartis sur une région de 580 kilobases incluant les gènes ARG1, CRSP3, ENPP3, ENPP1 et CTGF.

Les cercles bleus correspondent au - log10 (valeur de p) pour la comparaison des 298 individus témoins et des 421 enfants obèses. Les cercles rouges correspondent au - log10 (valeur de p) pour la comparaison des 298 individus témoins et de 62 enfants obèses issus des familles avec évidence de liaison au chromosome 6q. La figure n’est pas à l’échelle de la carte physique, et les cercles sont équidistants pour une meilleure visualisation.
Seuls, les polymorphismes IVS20 delT - 11 et A > G + 1044 TGA avaient un effet indépendant du K121Q sur la susceptibilité à l’obésité. L’haplotype à risque maximal d’obésité sévère, Q121, IVS20 delT - 11 et G + 1044 TGA, est présent chez 8 % de la population générale. Il augmente de 70 % le risque de développer une obésité infantile, de 37 % le risque d’obésité modérée et de 50 % le risque d’obésité morbide chez l’adulte. Cet haplotype à risque contribue partiellement à la liaison génétique observée au chromosome 6q, ce qui suggère l’implication probable d’autres gènes d’obésité dans cette région (Figure 4).
Figure 4
Liaison génétique avec l’obésité infantile avant et après exclusion des 15 paires de germains obèses partageant l’haplotype à risque.

Au maximum du pic, le Lod-score passe de 4,06 à 1,6, suggérant une contribution des SNP d’ENPP1 dans la liaison génétique.
L’haplotype à risque module la fonction et l’expression d’ENPP1
La mutation codante Q121 augmente l’activité inhibitrice du récepteur de l’insuline de la protéine ENPP1, sans moduler les niveaux d’ARNm ou de la protéine [16]. Comme les concentrations plasmatiques d’ENPP1 reflètent les niveaux tissulaires de ce peptide [19], ENPP1 a été dosée chez des enfants de poids variable. Les concentrations d’ENPP1 circulant étaient positivement corrélées à la corpulence et les enfants ayant une copie de l’haplotype à risque avaient des taux d’ENPP1 augmentés (Figure 5). Ainsi, les trois variants de ce gène, quand ils sont présents simultanément, modifieraient de manière complémentaire l’expression et la fonction de la protéine pour la rendre plus « insulinorésistante ».
Figure 5
Dosage plasmatique d’ENPP1 circulant chez 279 enfants.

A. Les niveaux d’ENPP1 circulant sont positivement corrélés à la corpulence, estimée par le score Z d’indice de masse corporelle (coefficient de corrélation de Pearson = 0,1, p = 0,05). B. Chez les enfants minces, la présence d’au moins une copie de l’haplotype à risque était associée à une augmentation de 19 % des niveaux circulants d’ENPP1 (p = 0,008).
ENPP1 est associé à une altération de l’homéostasie glucidique
Les enfants porteurs de l’haplotype à risque d’obésité ont une glycémie à jeun plus élevée, et un risque multiplié par 3,4 de développer une intolérance au glucose ou un DT2. Chez leurs parents porteurs du même haplotype, le risque de DT2 est globalement multiplié par 2,3. Cependant, en présence d’obésité, le risque est multiplié par 3,3, suggérant que ce gène favorise la survenue de diabète en présence d’un surpoids. Cette association avec le DT2 a ensuite été confirmée chez 2 500 sujets diabétiques et témoins d’origine européenne. ENNP1 est ubiquitaire, ce qui ne facilite pas la compréhension du phénotype de « diabésité » associé à ses variants géniques. Cependant, l’analyse par RT-PCR de l’expression des différentes isoformes de ce gène dans les principaux tissus humains a montré que le transcrit incluant 1 170 bases de la région en 3’UTR (et incluant le polymorphisme A > G + 1044 associé à l’obésité) était uniquement exprimé dans le foie, l’adipocyte et les îlots β-pancréatiques, trois tissus clés de l’homéostasie glucidique.
Vers la dissection génétique des obésités
L’identification d’ENPP1 comme gène de « diabésité » donne une première base moléculaire à l’hypothèse de l’hétérogénéité physiopathologique du trait complexe obésité. Tous les obèses ne sont pas très insulinorésistants et tous n’ont pas un syndrome métabolique sévère conduisant à un risque élevé de DT2 et de maladies cardio-vasculaires. La présence de variants d’ENPP1 favoriserait le développement dès l’enfance d’un état d’insulinorésistance chronique dont l’origine est évidemment multifactorielle. D’autres gènes sont probablement en cause qui pourraient intervenir dans les voies de signalisation de l’insuline, voire en amont dans les mécanismes de contrôle nerveux de la thermogenèse ou en aval au niveau mitochondrial. L’environnement joue un rôle crucial par l’alimentation riche en graisses à acides à longue chaîne de type oméga 6 ou en glucides à haut indice glycémique ou par la sédentarité qui exacerbe l’insulinorésistance. Cette obésité à fort impact délétère métabolique s’accompagne d’infarctus du myocarde précoces chez des diabétiques porteurs de la mutation Q121 d’ENPP1 [20].
Les résultats actuels, obtenus à partir de l’étude de plus de 6 000 personnes [21], montrent qu’au-delà des perturbations de la prise alimentaire et de la satiété [22-25], il existe des obésités d’origine « métabolique ». Ces obésités devront être prévenues ou prises en charge de manière différente que celles liées à des troubles du comportement alimentaire. Ainsi, des régimes alimentaires moins insulinorésistants, une activité physique importante, voire des médicaments spécifiques pourraient être très efficaces pour lutter contre la pandémie de diabésité qui est en train de submerger le monde du XXIe siècle.
Parties annexes
Références
- 1. Ebbeling CB, Pawlak, D. Ludwig DS. Childhood obesity : public health crisis, common sense cure. Lancet 2002 ; 360 : 473-82.
- 2. ObEpi. ObEpi 2003 : 3e enquête épidémiologique nationale sur l’obésité et le surpoids en France. Conférence de presse, Hôtel Dieu-Paris, 2003.
- 3. Stunkard AJ, Foch TT, Hrubec Z. A twin study of human obesity. JAMA 1986 ; 256 : 51-4
- 4. Mokdad AH, Ford ES, Bowman BA, et al. Prevalence of obesity, diabetes, and obesity-related health risk factors. JAMA 2003 ; 289 : 76-9.
- 5. Weill J, Vanderbecken S, Froguel P. Understanding the rising incidence of type 2 diabetes in adolescence. Arch Dis Child 2004 ; 89 : 502-4.
- 6. Meyre D, Lecoeur C, Delplanque J, et al. A genome-wide scan for childhood obesity-associated traits in French families shows significant linkage on chromosome 6q22.31-q23.2. Diabetes 2004 ; 53 : 803-11.
- 7. Atwood LD, Heard-Costa NL, Cupples LA, et al. Genomewide linkage analysis of body mass index across 28 years of the Framingham heart study. Am J Hum Genet 2002 ; 71 : 1044-50.
- 8. Duggirala R, Blangero J, Almasy L, et al. A major locus for fasting insulin concentrations and insulin resistance on chromosome 6q with strong pleiotropic effects on obesity-related phenotypes in nondiabetic Mexican Americans. Am J Hum Genet 2001 ; 68 : 1149-64.
- 9. Abney M, Ober C, McPeek MS. Quantitative-trait homozygosity and association mapping and empirical genomewide significance in large, complex pedigrees : fasting serum- insulin level in the Hutterites. Am J Hum Genet 2002 : 70 : 920-34.
- 10. Ehm MG, Karnoub MC, Sakul H, et al. Genomewide search for type 2 diabetes susceptibility genes in four American populations. Am J Hum Genet 2000 ; 66 : 1871-81.
- 11. Ghosh S, Watanabe RM, Valle TT, et al. The Finland-United States investigation of non-insulin-dependent diabetes mellitus genetics (FUSION) study. I. An autosomal genome scan for genes that predispose to type 2 diabetes. Am J Hum Genet 2000 ; 67 : 1174-85.
- 12. Demenais F, Kanninen T, Lindgren CM, et al. A meta-analysis of four European genome screens (GIFT consortium) shows evidence for a novel region on chromosome 17p11.2-q22 linked to type 2 diabetes. Hum Mol Genet 2003 ; 12 : 1865-73.
- 13. Xiang K, Wang Y, Zheng T, et al. Genome-wide search for type 2 diabetes/impaired glucose homeostasis susceptibility genes in the Chinese : significant linkage to chromosome 6q21-q23 and chromosome 1q21-q24. Diabetes 2004 ; 53 : 228-34.
- 14. Maddux BA, Sbraccia P, Kumakura S, et al. Membrane glycoprotein PC-1 and insulin resistance in non-insulin- dependent diabetes mellitus. Nature 1995 ; 373 : 448-51.
- 15. Maddux BA, Goldfine ID. Membrane glycoprotein PC-1 inhibition of insulin receptor function occurs via direct interaction with the receptor alpha-subunit. Diabetes 2000 ; 49 : 13-9.
- 16. Costanzo BV, Trischitta V, Di Paola R, et al. The Q allele variant (GLN121) of membrane glycoprotein PC-1 interacts with the insulin receptor and inhibits insulin signaling more effectively than the common K allele variant (LYS121). Diabetes 2001 ; 50 : 831-6.
- 17. Pizzuti A, Frittitta L, Argiolas A, et al. A polymorphism (K121Q) of the human glycoprotein PC-1 gene coding region is strongly associated with insulin resistance. Diabetes 1999 ; 48 : 1881-4.
- 18. Abate N, Carulli L, Cabo-Chan A, et al. Genetic polymorphism PC-1 K121Q and ethnic susceptibility to insulin resistance. J Clin Endocrinol Metab 2003 ; 88 : 5927-34.
- 19. Rutsch F, Vaingankar S, Johnson K, et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol 2001 ; 158 : 543-54.
- 20. Bacci S, Ludovico O, Prudente S, et al. The K121Q polymorphism of the ENPP1/PC-1 gene is associated with insulin resistance/atherogenic phenotypes, including earlier onset of type 2 diabetes and myocardial infarction. Diabetes 2005 ; 54 : 3021-5.
- 21. Meyre D, Bouatia-Naji N, Tounian A, et al. Variants of ENPP1 are associated with childhood and adult obesity and increase the risk of glucose intolerance and type 2 diabetes. Nat Genet 2005 ; 37 : 863-7.
- 22. Clement K, Vaisse C, Lahlou N, et al. A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction. Nature 1998 ; 392 : 398-401.
- 23. Vaisse C, Clement K, Guy-Grand B, Froguel PA. Frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat Genet 1998 ; 20 : 113-4.
- 24. Vaisse C, Clement K, Durand E, et al. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 2000 ; 10 : 253-62.
- 25. Boutin P, Dina C, Vasseur F, et al. GAD2 on chromosome 10p12 is a candidate gene for human obesity. PLOS Biol 2003 ; 1 : 1-11.
Liste des figures
Figure 1
Un intervalle de 2,4 Mb commun à huit études génome entier de « diabésité ».
Figure 2
Carte de déséquilibre de liaison pour 53 SNP répartis sur une région de 580 kilobases incluant les gènes ARG1, CRSP3, ENPP3, ENPP1 et CTGF.
Le génotypage des 53 SNP a été réalisé chez 298 individus témoins et 421 enfants obèses. Les régions de haut et de bas déséquilibre de liaison (indice delta) sont représentées respectivement par les couleurs rouges et bleues. La carte est à l’échelle physique.
Figure 3
Association des 53 SNP répartis sur une région de 580 kilobases incluant les gènes ARG1, CRSP3, ENPP3, ENPP1 et CTGF.
Les cercles bleus correspondent au - log10 (valeur de p) pour la comparaison des 298 individus témoins et des 421 enfants obèses. Les cercles rouges correspondent au - log10 (valeur de p) pour la comparaison des 298 individus témoins et de 62 enfants obèses issus des familles avec évidence de liaison au chromosome 6q. La figure n’est pas à l’échelle de la carte physique, et les cercles sont équidistants pour une meilleure visualisation.
Figure 4
Liaison génétique avec l’obésité infantile avant et après exclusion des 15 paires de germains obèses partageant l’haplotype à risque.
Au maximum du pic, le Lod-score passe de 4,06 à 1,6, suggérant une contribution des SNP d’ENPP1 dans la liaison génétique.
Figure 5
Dosage plasmatique d’ENPP1 circulant chez 279 enfants.
A. Les niveaux d’ENPP1 circulant sont positivement corrélés à la corpulence, estimée par le score Z d’indice de masse corporelle (coefficient de corrélation de Pearson = 0,1, p = 0,05). B. Chez les enfants minces, la présence d’au moins une copie de l’haplotype à risque était associée à une augmentation de 19 % des niveaux circulants d’ENPP1 (p = 0,008).