Résumés
Résumé
Longtemps considérée comme une maladie unique, l'amylose est aujourd'hui reconnue comme la marque histologique d'un ensemble de maladies, les amyloses. L'amylose est la voie finale commune, chez l'homme et dans de nombreuses espèces animales, de l'agrégation pathologique de plus de vingt protéines appartenant à des familles dénuées de relation fonctionnelle ou structurale. Les mécanismes de formation de ces agrégats commencent à être mieux connus. L'étape centrale, que l'on peut artificiellement reproduire in vitro, est un changement de conformation d'une protéine native en une protéine apte à l'auto-agrégation, sous une forme essentiellement formée de feuillets β. Le traitement actuel des amyloses, qui consiste à réduire la disponibilité en protéine amyloïde, n'est pas pleinement satisfaisant. La reconnaissance progressive des différentes étapes de ce phénomène pathologique a conduit à la conception de cibles thérapeutiques nouvelles : stabilisation de la protéine native, désagrégation des structures déjà β-plissées ou, encore, inhibition des liaisons avec certains composants du tissu conjonctif. Différentes approches, pharmacologiques et immunologiques, sont à l'étude sur des systèmes cellulaires et des modèles animaux, et certaines molécules sont parvenues au stade de l'essai clinique chez l'homme.
Summary
Amyloidosis bears many characteristics of orphan diseases. Its diagnosis is difficult and often delayed. The main reasons thereof are its quite various clinical presentation: amyloidosis behaves as a new great masquerader, and the need to get a tissue sample to submit to specific dyes. Although we have been able for a long time to recognize amyloid, its intimate nature has remained quite completely enigmatic until recently. In fact, major advances in this way have appeared only in the last decade and it is now possible to consider the mechanisms of amyloidosis as a multistep phenomenon. Amyloidosis is no more thought only as a « storage disease » of the extracellular space. This archaic viewpoint has shifted to the emerging paradigm of misfolded protein disorders. Amyloid proteins thus appear as a subgroup of misfolded proteins, where misfolding leads to subsequent aggregation. This aggregation may be a generic property of polypeptide chains possibly linked to their common peptide backbone that does not depend on specific amino acid sequences. And, in fact, many proteins can in vitro form amyloid-like aggregates, while in vivo, only 20 amyloid proteins have been so far identified. Although misfolding and aggregation are quite well studied in vitro, the last step of amyloid deposition, i.e. anchorage to the extracellular matrix, can not be so easily approached. Proteoglycans and serum amyloid P component have nevertheless been identified as key elements involved in extracellular deposition of amyloid proteins. These advances have opened new avenues in the therapeutic of amyloid disorders. Current treatment consists of support or replacement of impaired organ function and measures to reduce the production of amyloidogenic precursor proteins. Potential novel therapeutic strategies include stabilisation of the native fold of precursor proteins with targeted small molecules, reversion of misfolded proteins to their native state with « beta-sheet breakers », inhibition of amyloid fibril propagation and enhancement of amyloid clearance either through immunotherapy or by reducing the stability of deposits through depletion of serum amyloid P component, and breaking the anchorage to the extracellular matrix with glycosaminoglycan analogs.
Corps de l’article
C'est l'anatomopathologiste berlinois Rudolf Virchow qui, au xixe siècle, a créé le terme « amylose » pour désigner une substance, présente dans les tissus animaux, ayant des affinités tinctoriales communes avec l'amidon [1]. L'amylose, ou amyloïdose, désigne en français la lésion histologique ou substance (dépôt) amyloïde aussi bien que la maladie, alors que l'anglais dispose de deux mots : amyloid pour la lésion et amyloidosis pour la maladie. Naguère, l'amylose était considérée comme une des maladies de surcharge de l'espace extracellulaire, dont le caractère irréversible semblait bien établi. La nature intime de la substance amyloïde, bien que reconnue rapidement comme essentiellement protidique et peu glucidique, est restée longtemps inconnue. Ce n'est que récemment que l'analyse biochimique a permis de découvrir la diversité des protéines impliquées in vivo, et ce démembrement n'est pas encore totalement achevé (Tableau I). En outre, des agrégats intracellulaires très proches de l'agrégat amyloïde ont été identifiés dans plusieurs maladies du système nerveux central (maladie de Parkinson, maladies à prion, maladie d'Alzheimer), où il existe aussi pour la dernière, stricto sensu, des lésions d'amylose. Ces agrégats apparaissent maintenant comme une des conséquences possibles d'un mauvais repliement des protéines. L'amylose, maladie de surcharge, est donc devenue une des formes de maladie du repliement des protéines.
Tableau I
Nomenclature et classification des amyloses.

G : amylose généralisée ; L : amylose localisée ; Préc : précurseur ; Nomenclature préliminaire.
Les dépôts d'amylose peuvent se constituer quasiment dans tous les tissus et organes, où ils occupent de l'espace : il en résulte souvent une organomégalie. Il n'y a toutefois pas de signes cliniques véritablement spécifiques de l'amylose pour le clinicien. Les conséquences de l'infiltration amyloïde sont une défaillance progressive du fonctionnement des organes, et l'importance des lésions vasculaires rend compte de la fréquence des lésions ischémiques et hémorragiques. S'il n'est pas utile de développer ici l'ensemble des manifestations cliniques de l'amylose [2, 3], il faut toutefois mentionner les difficultés du diagnostic de cette maladie liées non seulement à sa rareté et au fait qu'elle peut simuler de nombreuses autres affections, mais aussi au critère diagnostique qui, puisqu'il est histologique, nécessite la réalisation d'une biopsie.
La substance amyloïde : du rouge Congo aux composants biochimiques
L'amylose, qu'elle soit localisée ou généralisée, forme au contact des matrices extracellulaires (membranes basales, tissu conjonctif interstitiel ou intercellulaire, parois vasculaires) des dépôts extracellulaires amorphes, acellulaires, craquelés et faiblement chromophiles, d'aspect « délavé ». Si le diagnostic de dépôt amyloïde peut être parfois envisagé sur les colorations tissulaires standard, la reconnaissance en microscopie optique de la nature amyloïde des dépôts requiert l'observation en lumière polarisée d'une coupe tissulaire fixée ou congelée colorée au rouge Congo : les dépôts d'amylose se colorent alors en rouge brique (congophilie) (Figure 1A). Cette fixation n'est pas suffisante pour identifier un dépôt amyloïde, car certains collagènes fixent également le rouge Congo. En revanche, la biréfringence jaune-vert en lumière polarisée qu'offre cette substance colorée par le rouge Congo (Figure 1B) est caractéristique, sinon spécifique, de l'amylose. Elle proviendrait de la liaison du colorant aux feuillets β plissés de la fibrille amyloïde [4]. D'autres colorations utilisées, rouge Sirius, thioflavine T et cristal violet, n'ont pas la spécificité de la coloration au rouge Congo.
Figure 1
Goitre amyloïde au cours d'une amylose AA.
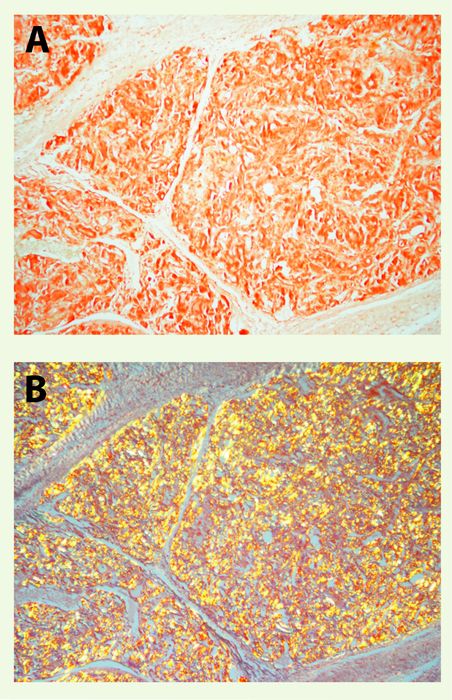
Dépôts amyloïdes massifs colorés en rouge brique (congophilie) (A) (x250) et biréfringence jaune-vert caractéristique en lumière polarisée (B).
L'étude ultrastructurale peut se révéler nécessaire lorsque les dépôts d'amylose sont trop petits pour être visibles en microscopie optique. Elle permet d'observer directement les fibrilles amyloïdes, qui ont un aspect ultrastructural caractéristique et unique, commun à toutes les variétés d'amylose. Ces fibrilles mesurent entre 75 et 100 µÅ de diamètre, mais leur longueur reste encore indéterminée. Elles sont enchevêtrées, mais non branchées les unes aux autres, rappelant l'aspect d'un paquet d'épingles jeté à terre. Ces fibrilles sont constituées de l'agrégation d'une protéine caractéristique d'un dépôt donné, appelée en conséquence protéine amyloïde. Cette agrégation s'effectue de façon caractéristique, sous forme de feuillets β qui offrent en diffraction aux rayons X un aspect typique. Cette technique ultrastructurale ne s'applique cependant qu'à des agrégats obtenus in vitro ou ex vivo, et reste du domaine de la recherche et non du diagnostic.
L'immuno-histochimie est la technique actuelle de référence pour la caractérisation de la protéine amyloïde. Elle permet d'étudier la fixation, sur les dépôts d'amylose, d'anticorps monoclonaux ou polyclonaux dirigés contre la majorité des protéines amyloïdes connues. Cette technique doit être réalisée sur un prélèvement tissulaire congelé. La mise en évidence de la chaîne légère d'immunoglobulines κ ou λ, définissant l'amylose AL, est très difficile sur une coupe tissulaire fixée, et comporte ainsi un risque de résultat faussement négatif.
De façon plus récente, plusieurs microméthodes biochimiques d'extraction des protéines amyloïdes directement à partir des dépôts d'amylose ont été mises au point. Elles permettent une caractérisation très précise de la protéine amyloïde par une analyse de sa séquence en acides aminés [5]. Dans la pratique, cette approche est cependant complexe et lourde. Pour les formes héréditaires des amyloses, à transmission autosomique dominante, le diagnostic génétique est guidé par la clinique et l'immuno-histochimie [3].
Si la substance amyloïde est composée à 80 % environ d'une protéine majoritaire, le reste comprend une variété de composants systématiquement associés, tels que l'ubiquitaire composant amyloïde P. Cette glycoprotéine appartient à la famille des pentraxines, qui contient aussi la protéine C réactive [6]. Un dérivé radiomarqué du composant P est utilisé pour localiser les dépôts amyloïdes par scintigraphie : cette technique, précieuse pour apprécier l'étendue des dépôts, n'est fonctionnelle en routine que dans deux pays européens [7]. En dehors de sa présence dans tous les dépôts amyloïdes, la fonction du composant P reste à élucider ; une hypothèse récente lui confère un rôle de protéine chaperon extracellulaire. D'autres composants ont également été identifiés dans les dépôts : des héparanes sulfates protéoglycanes, l'apolipoprotéine E (liant du Zn, notamment dans la maladie d'Alzheimer) et l'amyloid enhancing factor. Enfin, de nombreuses études mentionnent la présence de métaux (Cu, Zn ou Fe) associés aux dépôts.
Du repliement à l'agrégat spécifique
Les protéines précurseurs des polypeptides retrouvés dans les dépôts sont, pour la plupart, des protéines circulantes parfaitement solubles. Par un mécanisme encore inconnu, ces polypeptides subissent une modification de leur repliement qui conduit à une structure secondaire majoritairement constituée de feuillets β plissés, responsables de leur insolubilité et de leur dépôt sous forme de fibrilles, quelle que soit leur structure de départ (Figure 2).
Figure 2
Mécanismes des amyloses.

Le précurseur protéique est l'objet de modifications quantitatives ou qualitatives dans un contexte pathologique variable (inflammation, prolifération cellulaire, vieillissement), et change de conformation spatiale. Certains composés, de conformation intermédiaire entre la protéine native et la forme dénaturée, ont une structure instable propice à l'auto-agrégation. Au cours de ce phénomène apparaissent des agrégats de taille variable, du dimère au macro-agrégat qu'est la fibrille amyloïde. L'agrégat responsable de la toxicité cellulaire n'est pas identifié avec certitude. Les différentes structures peuvent être reconnues par des anticorps spécifiques de chaque étape [33] : certains anticorps reconnaissent d'ailleurs des épitopes communs à des oligomères de protéines amyloïdes différentes, en accord avec l'hypothèse d'une structure spatiale commune à ces agrégats [34]. L'immunologie ne se limite pas à l'aide au diagnostic (immuno-histochimie) et à la dissection des mécanismes de la maladie, mais pourrait participer à la thérapeutique [35].
En effet, si quelques protéines amyloïdes, comme la transthyrétine ou les chaînes légères des immunoglobulines, sont très riches en feuillets β plissés, d'autres, comme l'apolipoprotéine AI, sont majoritairement constituées d'hélices α Ces précurseurs n'ont donc pas de caractéristiques communes de structure, ni de fonction [8] ; il faut toutefois noter que trois d'entre elles sont des apolipoprotéines (AA, ApoAI et ApoAII). Le processus qui mène des protéines circulantes vers l'insolubilisation dans un dépôt amyloïde reste donc à éclaircir.
Parmi la vingtaine de protéines amyloïdes répertoriées jusque-là, celles impliquées dans certaines formes d'amyloses héréditaires, et qui sont l'objet d'une mutation ponctuelle, ont été surexprimées afin de comparer leur structure tridimensionnelle avec celles des protéines sauvages. Les résultats montrent peu de différence entre les mutants et les protéines sauvages, à l'exception d'une stabilité thermique parfois légèrement diminuée, donnant naissance à des composés intermédiaires à partir desquels se formeraient les agrégats [9]. Dans le cas de la transthyrétine, c'est le tétramère lui-même qui est déstabilisé par les mutations, tandis que la structure du monomère, siège de la mutation, est peu modifiée [10]. Les protéines amyloïdes isolées des dépôts sont aussi bien des protéines entières (lysozyme, insuline) que des fragments du précurseur. Dans ce dernier cas, le précurseur doit subir une protéolyse, sans que le stade auquel elle intervient ait pu être déterminé : une protéine mal repliée pourrait subir ce début de dégradation avant la formation des fibrilles, tout comme après.
Le repliement des protéines n'étant pas un processus au rendement parfait, plusieurs moyens existent, naturellement, pour réparer ou éliminer la protéine mal repliée. Dans la mesure où la probabilité d'échecs de repliement est plus importante dans le cas de protéines mutées, la concentration de telles protéines mal repliées est suceptible d'augmenter dans l'organisme. Quant à la la cinétique de croissance des dépôts in vitro, elle comporte, pour la plupart des protéines étudiées, une première étape de nucléation, puis une étape de croissance rapide, comme cela a été décrit dans le modèle historique de l'agrégation de l'hémoglobine S dans la drépanocytose [11]. L'agrégation d'une protéine amyloïde donnée peut même être accélérée par une protéine amyloïde hétérologue in vitro [12]. Toutefois, la croissance des agrégats pourrait différer selon le type de protéine amyloïde : in vitro, les agrégats de transthyrétine, notamment, ont une croissance linéaire dans certaines conditions [13].
La grande majorité des données nouvelles concernent la fibrillogenèse in vitro, qui permet de reproduire certaines caractéristiques de son équivalent in vivo. Ainsi, les fibrilles préparées in vitro présentent des caractéristiques semblables à celles isolées de dépôts amyloïdes : taille et aptitude à la biréfringence après coloration. De nombreuses études réalisées in vitro ont été dédiées à la formation de fibres à partir d'une large gamme de protéines, la plupart sans lien avec les protéines amyloïdes humaines, en les exposant à des conditions extrêmes de pH, de température ou d'additifs dénaturants. Des pistes récentes suggèrent l'implication de composés non encore identifiés, tels que des partenaires de la famille grandissante des intrinsically unfolded proteins. Ces protéines, dont le premier membre a été découvert il y a une dizaine d'années, sont naturellement non repliées et résistantes à une température élevée ; de plus, elles reposent la question fondamentale de la relation structure-fonction puisqu'elles sont actives dans leur état non replié [14]. Certaines protéines amyloïdes in vitro ont les caractéristiques de ces protéines intrinsèquement mal repliées [15]. La pertinence de ces expériences, réalisées dans des conditions éloignées de la physiologie, est discutable. En outre, il est particulièrement difficile d'étudier in vitro l'interaction des principaux composants (composant P, protéoglycanes) associés in vivo à la protéine amyloïde.
Certaines questions restent à ce jour très ouvertes. Il en est ainsi de la spécificité tissulaire du dépôt, dont on sait qu'elle dépend, en partie seulement, de la nature de la protéine amyloïde, mais probablement aussi de la spécificité très locale des protéines et glycoprotéines de la matrice extracellulaire. Les mécanismes par lesquels le vieillissement favorise l'émergence de dépôts amyloïdes (avec ou sans conséquence clinique) doit également être élucidé. Enfin, existe-t-il des formes d'amylose transmissibles, comme l'est le prion ? Des données suggèrent qu'au moins deux variétés d'amylose, l'amylose « sénile » de la souris, formée d'apolipoprotéine AII, et l'amylose AA de la souris peuvent être transmises dans certaines conditions expérimentales [16, 17].
Des mécanismes de la toxicité aux perspectives thérapeutiques
Les dépôts amyloïdes sont-ils d'innocents agrégats ?
Les mécanismes de la toxicité des dépôts amyloïdes sur les organes, tissus et cellules sont incomplètement élucidés. La vision classique d'une maladie de surcharge « étouffant » les organes massivement infiltrés par les dépôts amyloïdes ne peut plus être retenue aussi simplement : la toxicité des dépôts ne semble en effet pas proportionnelle à leur étendue. Il est ainsi frappant de constater que le foie peut contenir une masse supérieure à 1 000 g d'amylose sans aucun retentissement sur les fonctions cellulaires ou vasculaires hépatiques [1]. À l'opposé, les fonctions du nerf périphérique peuvent être sérieusement altérées alors que l'examen méticuleux de la biopsie nerveuse ne met en évidence que de discrets dépôts [1]. On peut légitimement déduire de ces données simples qu'il existe une sensibilité cellulaire variable d'un tissu à l'autre, ou que la toxicité, indépendante des dépôts, s'exerce par un autre mécanisme.
Cette question, qui n'était naguère même pas posée à propos des amyloses multisystémiques, est très curieusement au centre du débat autour de la maladie d'Alzheimer. Dans cette maladie, la toxicité neuronale des plaques séniles amyloïdes, formées par l'agrégation du peptide Aβ, est mise en balance avec une toxicité directe de ce dernier sur le neurone et avec le rôle primordial des agrégats de protéine tau à l'origine de la dégénérescence neurofibrillaire [18, 19]. Cette question est également posée pour les autres maladies du système nerveux central qui s'accompagnent d'inclusions neuronales formées d'agrégats protéiques : ainsi, dans la maladie de Parkinson, les rôles respectifs des monomères, oligomères et structures de degré supérieur dans la toxicité neuronale sont discutés. Les résultats d'études récentes menées in vitro suggèrent que les oligomères ou des protofibrilles seraient ici les plus toxiques : ces oligomères pourraient modifier la conductance des couches lipidiques membranaires ou former des pores à l'intérieur de la membrane, créant un néocanal ionique toxique directement responsable de la toxicité cellulaire [20, 21].
Quelques travaux ont d'ores et déjà été conduits sur les mécanismes de la toxicité cellulaire des protéines impliquées dans les amyloses multisystémiques. Ainsi, les chaînes légères d'immunoglobuline entraînent in vitro une toxicité sur les cellules myocardiques [22].
Cibles thérapeutiques dans les amyloses
Trois grands principes gouvernent le traitement des amyloses multisystémiques : le traitement symptomatique des défaillances organiques peut aller qu'à la substitution de l'organe détruit, essentiellement dans le cas du rein ; la transplantation d'organe, essentiellement rénale, mais aussi cardiaque ou hépatique, peut être envisagée en fonction de la diffusion de la maladie et de la maîtrise de la maladie sous-jacente. Cette maîtrise repose actuellement sur la diminution, voire la suppression, du précurseur de la protéine amyloïde : le grand intérêt de cette stratégie est que les effets s'exercent quelle que soit la structure impliquée dans la toxicité, protéine amyloïde, oligomère ou agrégat de forte taille, et quels que soient les mécanismes de la toxicité. Ce principe résume actuellement la thérapeutique de l'amylose. La troisième piste est encore de l'ordre de la recherche. On peut maintenant concevoir de s'opposer à différentes phases de la formation de l'agrégat amyloïde et aux mécanismes de leur toxicité cellulaire.
Comment diminuer la disponibilité en protéine amyloïde ?
Si les thérapeutiques actuelles ont comme principal objectif d'annuler, ou au moins de diminuer, la production de la protéine amyloïde ou de son précurseur, d'autres options sont théoriquement envisageables : épuration plasmatique, augmentation de son catabolisme… Elles n'ont pas jusqu'à ce jour d'application clinique. Le traitement est donc directement dépendant des mécanismes en cause dans la production de la protéine toxique : traitement du clone plasmocytaire dans l'amylose AL [23], maîtrise de l'inflammation dans l'amylose AA, transplantation hépatique dans l'amylose héréditaire ATTR [24]. Ces traitements sont de plus en plus efficaces, mais de mise en oeuvre lourde, et grevés d'une toxicité souvent majeure.
Traitements du futur
Les progrès dans l'identification des différentes étapes de la formation des dépôts amyloïdes permettent d'envisager des traitements pharmacologiques ou immunitaires plus spécifiques de ces maladies. Nous n'évoquerons que certaines de ces nouvelles cibles. L'auto-agrégation de la transthyrétine, molécule homotétramérique, passe par la déstabilisation du tétramère et la formation d'un intermédiaire amylogène monomérique. La stabilité de la transthyrétine, obtenue naturellement par la fixation à son ligand, peut être maintenue in vitro par la fixation de petites molécules, dont une série d'analogues du diflunisal, un antalgique usuel [25]. Ces molécules sont actuellement en expérimentation chez des souris transgéniques TTR-V30M, c'est-à-dire modifiées par le gène de la transthyrétine, siège de la mutation V30M.
Il n'est plus inconcevable de pouvoir éliminer des tissus les dépôts déjà formés. L'I-DOX, une anthracycline utilisée contre le clone plasmocytaire, a fait la preuve spectaculaire de la possibilité d'éliminer des dépôts amyloïdes in vivo [26]. Son action restreinte à certains tissus n'a pas autorisé son développement clinique, mais sa valeur heuristique est certaine. In vitro, d'autres molécules moins toxiques, comme les antibiotiques de la famille des cyclines, peuvent « dissoudre » des agrégats de transthyrétine : ces molécules sont en cours d'essai chez les souris TTR-V30M [27].
La phase d'auto-agrégation des protéines amyloïdes est l'objet d'approches multiples. Des peptides « briseurs de feuillets β » ont été synthétisés afin de s'opposer à l'interaction entre deux protéines [28]. Cette approche est potentiellement limitée par la nature même de la molécule thérapeutique, un court peptide. Pour surmonter cette difficulté, une molécule bifonctionnelle a été récemment conçue, portant à une extrémité le colorant rouge Congo, et à l'autre un ligand d'une grosse protéine chaperon intracellulaire, la FKBP (FK506 binding protein). Le rouge Congo se lie aux feuillets β sans les modifier, et le ligand est harnaché à la protéine chaperon : in vitro, cette molécule a le plus puissant effet inhibiteur, à de faibles concentrations, sur la fibrillogenèse du peptide Aβ [29].
L'interaction avec des constituants naturels de la matrice extracellulaire (essentiellement les protéoglycanes et le composant amyloïde P) est in vivo une étape cruciale de la formation des dépôts amyloïdes. Dans un modèle murin d'amylose AA, de petites molécules de type sulfate et sulfonate ont été utilisées, avec succès, pour inhiber l'interaction entre l'héparane sulfate et la protéine amyloïde AA [30]. L'une d'entre elles est en expérimentation chez l'homme, dans un essai randomisé, tandis que d'autres composés sont en cours d'élaboration [31]. Pour le composant amyloïde P, l'idée consiste à épurer le composant P du plasma, prévenant ainsi son action pro-amylogène. Cette épuration a été obtenue grâce à de petites molécules qui relient deux pentamères de composant P, l'ensemble formé étant rapidement dégradé dans le plasma. L'une de ces molécules a fait l'objet d'un essai préliminaire chez l'homme [32].
Conclusions
Nos connaissances sur les amyloses ont considérablement progressé, aussi bien dans le domaine de l'analyse des composants de la substance amyloïde que des mécanismes de la fibillogenèse in vitro. De nombreuses inconnues demeurent toutefois, dans la nature exacte des phénomènes qui se produisent in vivo. Parallèlement, les données acquises ont conduit à une évolution majeure des concepts thérapeutiques, dont certains commencent à être appliqués chez l'homme.
Parties annexes
Références
- 1. Grateau G, Benson MD, Delpech M. Les amyloses. Paris : Flammarion Médecine-Sciences, 2000 : 580 p.
- 2. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses. N Engl J Med 1997 ; 337 : 898-909.
- 3. Grateau G. Amyloses. In : Kahn MF, Peltier O, Meyer O, Piette JC, eds. Les maladies systémiques. Paris : Flammarion Médecine-Sciences, 2000 : 1279-308.
- 4. Carter DB, Chou KC. A model for structure-dependent binding of Congo red to Alzheimer beta-amyloid fibrils. Neurobiol Aging 1998 ; 19: 37-40.
- 5. Kaplan B, Shtrasburg S, Pras M. Micropurification techniques in the analysis of amyloid proteins. J Clin Pathol 2003 ; 56 : 86-90.
- 6. Baltz ML, Caspi D, Evans DJ, et al. Circulating serum amyloid P component is the precursor of amyloid P component in tissue amyloid deposits. Clin Exp Immunol 1986 ; 66 : 691-700.
- 7. Hawkins PN. Serum amyloid P component scintigraphy for diagnosis and monitoring amyloidosis. Curr Opin Nephrol Hypertens 2002 ; 11 : 649-55.
- 8. Sipe JD, Ed. Amyloid proteins: the beta sheet conformation and disease. Weinheim: Wiley-VCH, 2005 (sous presse).
- 9. Canet D, Sunde M, Last AM, et al. Mechanistic studies of the folding of human lysozyme and the origin of amyloidogenic behavior in its disease-related variants. Biochemistry 1999 ; 38 : 6419-27.
- 10. Hamilton JA, Benson MD. Transthyretin: A review from a structural perspective. Cell Mol Life Sci 2001 ; 58 : 1-31.
- 11. Hofrichter J, Ross PD, Eaton WA. Kinetics and mechanism of deoxyhemoglobin S gelation: a new approach to understanding sickle cell disease. Proc Natl Acad Sci USA 1974 ; 71 : 4864-8.
- 12. O'Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem 2004 ; 279 : 17490-9.
- 13. Hurshman AR, White JT, Powers ET, Kelly JW. Transthyretin aggregation under partially denaturing conditions is a downhill polymerization. Biochemistry 2004 ; 43 : 7365-81.
- 14. Tompa P. Intrinsically unstructured proteins. Trends Biochem Sci 2002 ; 27 : 527-33.
- 15. Niraula TN, Konno T, Li H, et al. Pressure-dissociable reversible assembly of intrinsically denatured lysozyme is a precursor for amyloid fibrils. Proc Natl Acad Sci USA 2004 ; 101 : 4089-93.
- 16. Korenaga T, Fu X, Xing Y, et al. Tissue distribution, biochemical properties, and transmission of mouse Type A AApoAII amyloid fibrils. Am J Pathol 2004 ; 164 : 1597-606.
- 17. Lundmark K, Westermark GT, Nystrom S, et al. Transmissibility of systemic amyloidosis by a prion-like mechanism. Proc Natl Acad Sci USA 2002 ; 99 : 6979-84.
- 18. Mudher A, Lovestone S. Alzheimer's disease : do tauists and baptists finally shake hands? Trends Neurosci 2002 ; 25 : 22-6.
- 19. Lee HG, Casadesus G, Zhu X, et al. Challenging the amyloid cascade hypothesis: senile plaques and amyloid-beta as protective adaptations to Alzheimer disease. Ann NY Acad Sci 2004 ; 1019 : 1-4.
- 20. Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003 ; 300 : 486-9.
- 21. Kayed R, Sokolov Y, Edmonds B, et al. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem 2004 ; 279 : 46363-6.
- 22. Brenner DA, Jain M, Pimentel DR, et al. Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res 2004 ; 94 : 1008-10.
- 23. Skinner M, Sanchorawala V, Seldin DC, et al. High-dose melphalan and autologous stem-cell transplantation in patients with AL amyloidosis: an 8-year study. Ann Intern Med 2004 ; 140 : 85-93.
- 24. Stangou AJ, Hawkins PN. Liver transplantation in transthyretin-related familial amyloid polyneuropathy. Curr Opin Neurol 2004 ; 17 : 615-20.
- 25. Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem 2004 ; 47 : 355-74.
- 26. Gertz MA, Lacy MQ, Dispenzieri A, et al. A multicenter phase II trial of 4`-iodo-4`deoxydoxorubicin (IDOX) in primary amyloidosis (AL). Amyloid 2002 ; 9 : 24-30.
- 27. Cardoso I, Merlini G, Saraiva MJ. 4`-iodo-4`-deoxydoxorubicin and tetracyclines disrupt transthyretin amyloid fibrils in vitro producing noncytotoxic species: screening for TTR fibril disrupters. FASEB J 2003 ; 17 : 803-9.
- 28. Soto C, Kascsak RJ, Saborio GP, et al. Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet 2000 ; 355 : 192-7.
- 29. Gestwicki JE, Crabtree GR, Graef IA. Harnessing chaperones to generate small-molecule inhibitors of amyloid beta aggregation. Science 2004 ; 306 : 865-9.
- 30. Kisilevsky R, Lemieux LJ, Fraser PE, et al. Arresting amyloidosis in vivo using small-molecule anionic sulphonates or sulphates: implications for Alzheimer's disease. Nat Med 1995 ; 1 : 143-8.
- 31. Kisilevsky R, Szarek WA, Ancsin JB, et al. Inhibition of amyloid A amyloidogenesis in vivo and in tissue culture by 4-deoxy analogues of peracetylated 2-acetamido-2-deoxy-alpha- and beta-d-glucose: implications for the treatment of various amyloidoses. Am J Pathol 2004 ; 164 : 2127-37.
- 32. Pepys MB, Herbert J, Hutchinson WL. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature 2002 ; 417 : 254-9.
- 33. Dumoulin M, Dobson CM. Probing the origins, diagnosis and treatment of amyloid diseases using antibodies. Biochimie 2004 ; 86 : 589-600.
- 34. O'Nuallain B, Wetzel R. Conformational Abs recognizing a generic amyloid fibril epitope. Proc Natl Acad Sci USA 2002 ; 99 : 1485-90.
- 35. Hrncic R, Wall J, Wolfenbarger DA, et al. Antibody-mediated resolution of light chain-associated amyloid deposits. Am J Pathol 2000 ; 157 : 1239-46.
Liste des figures
Figure 1
Goitre amyloïde au cours d'une amylose AA.
Dépôts amyloïdes massifs colorés en rouge brique (congophilie) (A) (x250) et biréfringence jaune-vert caractéristique en lumière polarisée (B).
Figure 2
Mécanismes des amyloses.
Le précurseur protéique est l'objet de modifications quantitatives ou qualitatives dans un contexte pathologique variable (inflammation, prolifération cellulaire, vieillissement), et change de conformation spatiale. Certains composés, de conformation intermédiaire entre la protéine native et la forme dénaturée, ont une structure instable propice à l'auto-agrégation. Au cours de ce phénomène apparaissent des agrégats de taille variable, du dimère au macro-agrégat qu'est la fibrille amyloïde. L'agrégat responsable de la toxicité cellulaire n'est pas identifié avec certitude. Les différentes structures peuvent être reconnues par des anticorps spécifiques de chaque étape [33] : certains anticorps reconnaissent d'ailleurs des épitopes communs à des oligomères de protéines amyloïdes différentes, en accord avec l'hypothèse d'une structure spatiale commune à ces agrégats [34]. L'immunologie ne se limite pas à l'aide au diagnostic (immuno-histochimie) et à la dissection des mécanismes de la maladie, mais pourrait participer à la thérapeutique [35].
Liste des tableaux
Tableau I
Nomenclature et classification des amyloses.
G : amylose généralisée ; L : amylose localisée ; Préc : précurseur ; Nomenclature préliminaire.