
Résumés
Résumé
Le cancer de la vessie se présente sous deux formes différentes: la forme superficielle et la forme infiltrante. La forme superficielle se caractérise par une fréquence élevée de pertes (totales ou partielles) du chromosome 9, notamment du bras long. Ces observations suggèrent la présence de gènes suppresseurs de tumeur (GST) dans cette région. Nous présentons le gène PATCHED, localisé en position 9q22.3, déjà impliqué dans le carcinome basocellulaire de la peau, comme un GST potentiel dans le cancer superficiel de la vessie.
Summary
Superficial bladder cancer shows a high frequency of total or partial chromosome 9 losses. Loss of heterozygosity at position 9q22.3 is one of the most frequent and is associated with highly recurrent tumours. The PATCHED gene, ortholog of a gene first described in the drosophila as a segment polarity gene, is located at 9q22.3. It is a member of a signal transduction pathway and a tumour suppressor gene (TSG), involved in basal cell carcinoma. We propose PATCHED as a TSG candidate in superficial bladder cancer.
Corps de l’article
Le cancer de la vessie se situe au cinquième rang, en terme d’incidence, dans les sociétés occidentales. Son étiologie est principalement associée à la présence de produits carcinogènes dans l’environnement, notamment ceux de la fumée de cigarette et des émanations d’origine industrielle. Lors du diagnostic initial, 75 à 80% des patients présentent un cancer superficiel papillaire (stade TA ou T1) et pour plus de 60% de ces patients, l’évolution sera marquée par des récidives parfois fréquentes avec progression vers l’infiltration musculaire dans 10% des cas. Le reste des patients présentent un cancer d’emblée infiltrant (stade T2-T4) dérivant d’un carcinome in situ (CIS) de grade élevé et d’une dysplasie. Bien que la mortalité associée au cancer de la vessie soit due aux tumeurs infiltrantes, la morbidité provient principalement des tumeurs superficielles récidivantes et de leur traitement par résection transurétrale et chimiothérapie ou immunothérapie intravésicale.
Anomalies du chromosome 9 dans le cancer de la vessie
Les différentes expressions cliniques des cancers de la vessie ont pu être associées à des voies moléculaires distinctes [1]. Alors que l’on retrouve souvent des mutations du gène p53 dans les CIS et les tumeurs infiltrantes, des anomalies du chromosome 9 sont fréquentes dans les tumeurs superficielles. Diverses études ont montré des pertes d’hétérozygotie (LOH, loss ofheterozygosity) sur ce chromosome dans plus de la moitié des cancers de vessie et il s’agit souvent de l’unique altération génétique observée dans les tumeurs à un stade peu avancé. Ces données suggèrent la présence d’un ou plusieurs gènes suppresseur de tumeur (GST) sur le chromosome 9, intervenant dans les phases précoces de la cancérogenèse vésicale. Plusieurs études ont suggéré que les locus en 9p22-23, 9p21-22, 9p11-13, 9q12-13, 9q21-22, 9q31, 9q33 et 9q34 seraient susceptibles de porter des GST [1-4]. Lors d’une recherche de LOH au niveau de 28marqueurs microsatellites du chromosome 9 dans 139 tumeurs initiales de stade TA ou T1, nous avons répertorié 67 tumeurs (48%) présentant une ou plusieurs LOH sur le chromosome 9, ce qui nous a conduit à proposer 4 locus au niveau du bras 9q, 9q22.3, 9q31-32, 9q33 et 9q34 comme étant des sites potentiels de GST[2]. Quelques GST candidats ont été associés à certains locus du chromosome 9 tels CDKN2A à 9p21, PATCHED (PTC) à 9q12-31, DBCCR1 à 9q32-33 et TSC1 à 9q34 [1, 4]. La présence de LOH sur le chromosome 9 a de plus été associée à un risque élevé de récidive [5] et nous avons observé que des patients porteurs des tumeurs superficielles contenant des délétions dans les régions 9ptr-p22, 9q22.3, 9q33 et 9q34 avaient une survie sans récidive significativement plus courte que des patients dont les tumeurs étaient exemptes de délétions (Figure1) [6], ce qui est compatible avec la présence de GST au niveau de ces locus.
Dans le but d’identifier le GST de la région 9q22.3, nous avons poursuivi notre cartographie du chromosome 9 en analysant 11 marqueurs microsatellites additionnels dans cette région, sur 139 tumeurs de stade peu avancé [7]. Cette étude a permis de définir une région minimale de délétion de 0,5Mb incluant, le gène PTC, candidat GST précédemment proposé. Dans 46% de l’ensemble des tumeurs analysées, nous avons observé la perte d’un marqueur microsatellite situé dans l’intron 1 du gène PTC [7]. Ces données nous ont conduit à proposer que PTC soit effectivement un GST candidat de la région 9q22.3 dont la délétion serait un événement précoce de la cancérogenèse vésicale.
Voie de signalisation ptc/Hedgehog chez la drosophile
Le gène PTC humain est l’orthologue du gène ptc de la drosophile, d’abord identifié comme étant un gène important au cours du développement embryonnaire [8]. Chez la mouche, le gène ptc joue un rôle dans l’élaboration de l’axe antéro-postérieur des embryons et dans la polarité des segments. Il code pour une protéine possédant 12 domaines transmembranaires, deux boucles extracellulaires et un domaine intracellulaire. Cette protéine agit comme récepteur de la protéine sécrétée hedgehog (hh) (Figure 2). En l’absence de hh, ptc inhibe la voie de signalisation en formant un complexe avec une seconde protéine membranaire, smoothened (smo), et en réprimant l’activité de cette dernière. D’autres protéines engagées dans cette voie de signalisation ont également été décrites chez la drosophile, la protéine costal-2 (cos-2), la sérine-thréonine kinase fused (fu), le suppresseur de fused (su[fu]) et le facteur de transcription à doigts de zinc cubitusinterruptus (ci). Ces protéines forment un complexe cytoplasmique lié aux microtubules. Lorsque la voie est inactive, ci subit un clivage qui libère un fragment amino-terminal de 75 kDa. Ce fragment pénètre dans le noyau où il agit comme répresseur transcriptionnel. La liaison de hh à ptc libère smo, ce qui entraîne une dissociation du complexe cytoplasmique et l’inhibition du clivage de ci. La protéine ci intacte subit alors une maturation qui la transforme en un activateur transcriptionnel labile de divers gènes dont ptc, decapentaplegic (dpp), un membre de la famille TGFβ, et wingless (wg), orthologue des gènes Wnt des vertébrés [9].
Figure 1
Analyse de survie sans récidive par la méthode de Kaplan-Meier, selon la présence de délétions dans les régions 9ptr-p22, 9q22.3, 9q33, 9q34 et 9q34-qtr et pour le chromosome 9 entier.

La présence de délétions de marqueurs microsatellites est associée à un risque plus élevé de récidive dans les régions susceptibles de porter des GST bien qu’on ne retrouve aucune association ailleurs sur le chromosome (9q34-qtr). Le nombre de tumeurs dans chaque catégorie est indiqué au-dessus de la courbe; les cas non informatifs sont exclus de cette analyse. En bleu: courbe de survie avec LOH; en rouge: courbe de survie sans LOH (d’après [6]).
Figure 2
Modèle proposé pour la voie de signalisation ptc/hh chez la drosophile, d’après les données les plus récentes.
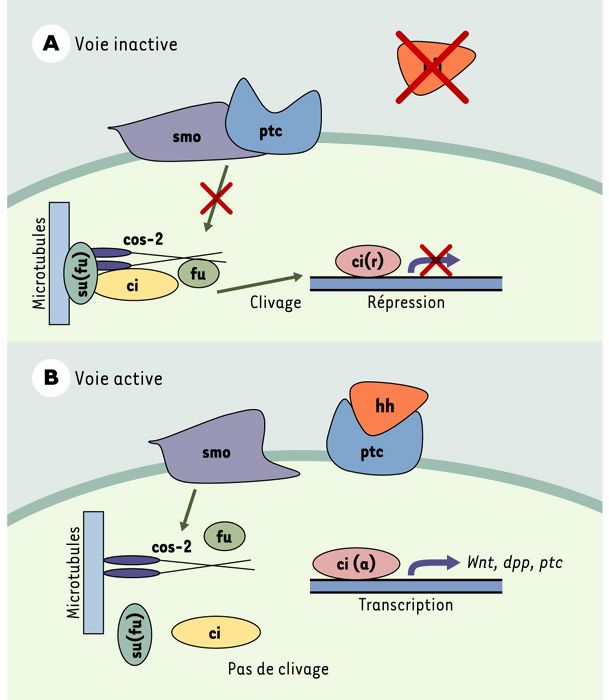
A. La voie est inactive lorsque ptc (Patched) se lie à la protéine smo (smoothered) et en inhibe l’activité. Un complexe de protéines cytoplasmiques liées aux microtubules amène le clivage de ci (facteur de transcription cubitus interruptus) en un répresseur transcriptionnel qui exerce son action dans le noyau. B. Voie active: la liaison du ligand hh (hedgedog) à ptc libère smo de l’action répressive de cette dernière, ce qui entraîne la dissociation du complexe cytoplasmique. ci ne subit pas de clivage et agit comme activateur de la transcription de divers gènes dont ptc lui-même. cos-2: protéine costal-2; fu: sérine-thréonine kinase fused; su(fu): suppresseur de fused.
Voie de signalisation PTC/SHH chez les vertébrés
Des homologues de plusieurs membres de cette voie ont été identifiés chez les vertébrés où celle-ci, bien que présentant un niveau plus élevé de complexité, semble fonctionner de façon similaire à celle de la drosophile [10, 11]. Deux homologues de ptc, PTC1 et PTC2, ont été décrits comme pouvant tous les deux interagir avec HH et smo [9, 11]. Cependant, bien que PTC1, dont le gène est situé sur le chromosome 9, soit bien caractérisé, on connaît peu le rôle de PTC2. Trois homologues de hh ont également été identifiés, Sonic (SHH), Indian (IHH) et Desert (DHH) Hedgehog [9]. Ces trois ligands utilisent la même voie de signalisation. Un seul homologue de smo a été identifié jusqu’à présent, mais trois facteurs de transcription à doigts de zinc, GLI-1, GLI-2 et GLI-3 occupent la position de ci [8, 12]. GLI-1 semble agir essentiellement comme activateur et serait le principal effecteur transcriptionnel de cette voie tandis que GLI-2 et GLI-3 possèdent des domaines activateurs et répresseurs de la transcription et sont modifiées par clivage protéolytique comme ci. Cependant, des incertitudes importantes subsistent quant à la transduction du signal SHH par les protéines GLI et la plupart des études suggèrent un équilibre complexe entre des fonctions divergentes. Une seule protéine cytoplasmique agissant en amont de GLI, l’homologue de su(fu), a été identifiée dans des cellules humaines [11].
Voie de signalisation PTC/SHH et prolifération cellulaire
Bien que les mécanismes sous-jacents au fonctionnement de la voie PTC/SHH demeurent encore mal compris, il apparaît que son activation se traduit principalement par une prolifération accrue. Le haut degré de conservation de cette voie chez la drosophile et les vertébrés permet de supposer que l’activation de la prolifération implique la participation des membres des familles TGFβ et WNT, ces dernières possédant un large spectre de fonctions. De plus, des données récentes ont permis d’établir un lien direct entre l’activation par hh et l’augmentation de la transcription des cyclines D et E, protéines directement engagées dans l’activation du cycle cellulaire [13]. Enfin, la transfection de cellules rénales de rat avec le gène Gli-1 entraîne des modifications du profil génique compatibles avec un accroissement de la prolifération par des voies diverses comme l’augmentation de la division cellulaire, la diminution de l’adhérence cellulaire et de l’apoptose. Des sites de reconnaissance de la protéine Gli-1 dans le promoteur d’au moins quatre des gènes modulés (cycline D2, IGFBP-6, osteopontine et plakoglobine) ont pu être identifiés [14].
Voie de signalisation PTC/SHH et cancer
Le rôle du gène PTC en tant que GST a été initialement démontré pour le carcinome basocellulaire de la peau [9]. En effet, des mutations du gène PTC sont fréquemment détectées, tant pour la forme familiale que pour la forme sporadique de ces carcinomes. En outre, comme pour les tumeurs superficielles de la vessie, on observe une perte d’hétérozygotie de marqueurs du locus PTC dans la région 9q dans environ 50% des carcinomes basocellulaires. La surexpression de GLI-1, ainsi que la présence de mutations dans les gènes SHH ou Smo peuvent aussi induire un carcinome basocellulaire, suggérant que toute anomalie menant à une surexpression de GLI-1 dans des cellules de la couche basale de la peau peut induire un carcinome [8, 9]. La capacité de GLI-1 à transformer des cellules en coopération avec le gène E1A de l’adénovirus a d’ailleurs confirmé sa nature oncogénique [12]. Des mutations des gènes PTC et Smo ont également été identifiées dans des médulloblastomes et des tumeurs primitives neuroectodermales [8]. Finalement, des souris porteuses d’un allèle inactivé du gène Ptc développent fréquemment des rhabdomyosarcomes [8].
Voie de signalisation PTC/SHH et cancer de la vessie
Il semble donc bien établi que des altérations de la voie PTC/SHH peuvent induire divers cancers. Les délétions fréquentes observées au locus 9q22 dans des tumeurs de la vessie par diverses équipes, dont la nôtre, amènent naturellement à suspecter le gène PTC comme un élément clé dans la progression conduisant au cancer de la vessie. Afin de vérifier cette hypothèse, la présence de mutations dans le gène PTC a été recherchée dans des tumeurs de la vessie. McGarvey et al. ont détecté des mutations du locus PTC dans deux tumeurs infiltrantes sur 54 tumeurs analysées [15]. Par ailleurs, dans cette étude, l’expression des gènes PTC, Smo et GLI-3 dans l’urothélium normal et dans des lignées cellulaires vésicales a permis de conclure que la voie PTC/SHH pourrait jouer un rôle dans le contrôle de la prolifération urothéliale et contribuerait à la genèse d’au moins un sous-groupe de carcinomes transitionnels de la vessie. En revanche, aucune mutation n’a été trouvée dans trois autres études [16-18]. Étant donné que le modèle initialement admis pour un GST suppose l’inactivation des deux allèles du gène, souvent par délétion d’une copie et mutation de l’autre, nous avons séquencé l’ADNc de 15 tumeurs TA ou T1 ayant perdu un allèle de PTC, sans trouver la moindre mutation [7]. La perte d’une copie d’un GST sans altération de l’autre copie ou haploinsuffisance, a été récemment rapportée comme suffisante pour promouvoir le cancer. Diverses études ont, en effet, rapporté l’haplo-insuffisance du gène PTEN dans le carcinome rénal à cellules claires [19], du gène SMAD4 dans des cancers gastriques [20], du gène p27 dans divers cancers [21] et du gène PTC lors de l’induction de médulloblastome chez la souris [22]. Nous avons analysé l’expression du gène PTC par la technique de RT-PCR semi-quantitative dans 21tumeurs superficielles de la vessie et 4 échantillons d’urothélium normal humain et avons observé une diminution significative de l’expression de ce gène dans les tumeurs ayant perdu une copie de PTC par rapport à son expression dans l’urothélium normal (Figure 3) [7]. Des fluctuations marquées de l’expression de PTC sont aussi constatées dans les tumeurs possédant les deux allèles de PTC suggérant que des mécanismes autre que l’haplo-insuffisance pourraient également contribuer à l’altération de l’expression de ce gène. Des données préliminaires suggèrent la diminution de l’expression d’autres gènes de la voie PTC/SHH.
Figure 3
Analyse de l’expression du gène PTC dans des tumeurs vésicales avec perte d’hétérozygotie (LOH) du marqueur PTC, sans LOH détectée sur le chromosome 9, et dans des urothéliums normaux (N).
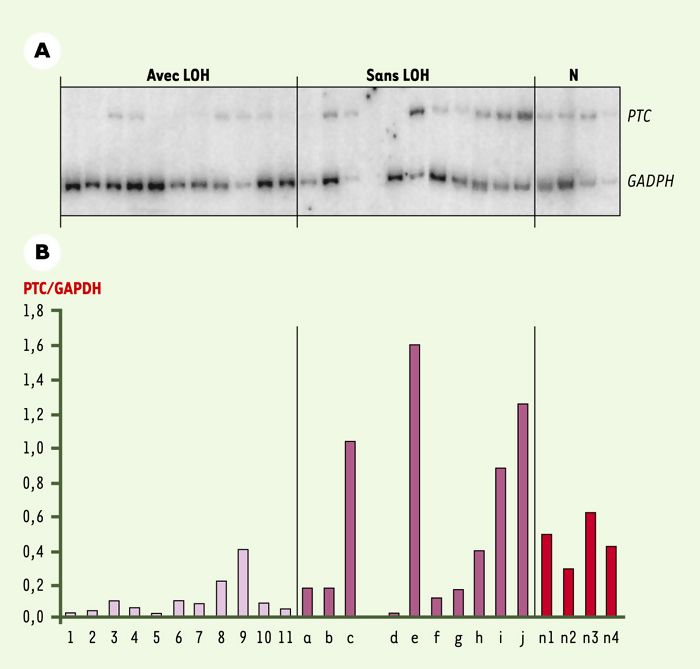
L’expression du gène a été analysée par la technique de RT-PCR semi-quantitative en présence d’un précurseur radioactif. A. Photographie d’un gel d’électrophorèse montrant la migration des fragments amplifiés pour le gène PTC et pour un gène témoin (glycéraldéhyde-3-phosphate déhydrogénase (GAPDH)). B. Histogramme montrant le rapport de l’intensité des bandes obtenues pour PTC et GAPDH pour les divers échantillons analysés (d’après [7]). On observe une diminution significative de l’expresion du gène dans les tumeurs ayant perdu une copie de PTC.
Conclusions
L’ensemble de ces données est compatible avec l’hypothèse de la perturbation de la voie de signalisation PTC/SHH au cours des phases précoces de la cancérogenèse vésicale, possiblement en rapport, dans environ la moitié des cas, avec la perte d’une copie du gène PTC. Les carcinomes basocellulaires de la peau et les carcinomes papillaires superficiels de la vessie partageant plusieurs caractéristiques comme une croissance locale, une induction par des carcinogènes environnementaux et un faible potentiel métastatique, il est plausible que des altérations génétiques semblables soient retrouvées dans ces deux types de cancers. Une étude plus approfondie des mécanismes en cause permettra éventuellement l’élaboration de thérapies permettant de ralentir et même de bloquer la prolifération des cellules cancéreuses dans la vessie.
Parties annexes
Références
- 1. Knowles MA. The genetics of transitional cell carcinoma: progress and potential clinical application. BJU Int 1999; 84: 412-27.
- 2. Simoneau M, Aboulkassim TO, Larue H, Rousseau F, Fradet Y. Four tumor suppressor loci on chromosome 9q in bladder cancer: evidence for two novel candidate regions at 9q22.3 and 9q31. Oncogene 1999; 18: 157-63.
- 3. Czerniak B, Chaturvedi V, Li L, et al. Superimposed histologic and genetic mapping of chromosome 9 in progression of human urinary bladder neoplasia: implications for a genetic model of multistep urothelial carcinogenesis and early detection of urinary bladder cancer. Oncogene 1999; 18: 1185-96.
- 4. Williams SV, Sibley KD, Davies AM, et al. Molecular genetic analysis of chromosome 9 candidate tumor-suppressor loci in bladder cancer cell lines. Genes Chrom Cancer 2002; 34: 86-96.
- 5. Bartlett JMS, Watters AD, Ballantyne SA, Going JJ, Grigor KM, Cooke TG. Is chromosome 9 loss a marker of disease recurrence in transitional cell carcinoma of the urinary bladder? Br J Cancer 1998; 77: 2193-8.
- 6. Simoneau M, Larue H, Aboulkassim TO, Meyer F, Moore L, Fradet Y. Chromosome 9 deletions and recurrence of superficial bladder cancer: identification of four regions of prognostic interest. Oncogene 2000; 19: 6317-23.
- 7. Aboulkassim TO, Larue H, Lemieux P, Rousseau F, Fradet Y. Alteration of the Patched locus in superficial bladder cancer. Oncogene 2003; 22: 2967-71.
- 8. Wicking C, Smyth I, Bale A. The hedgehog signalling pathway in tumorigenesis and development. Oncogene 1999; 18: 7844-51.
- 9. Bale AE, Yu KP. The hedgehog pathway and basal cell carcinomas. Hum Mol Genet 2001; 10: 757-62.
- 10. Goodrich LV, Johnson RL, Milenkovic L, McMahon JA, Scott MP. Conservation of the hedgehog/patched signaling pathway from flies to mice: induction of a mouse patched gene by Hedgehog. Genes Dev 1996; 10: 301-12.
- 11. Villavicencio EH, Walterhouse DO, Iannaccone PM. The sonic hedgehog-patched-gli pathway in human development and disease. Am J Hum Genet 2000; 67: 1047-54.
- 12. Altaba A. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development 1999; 126: 3205-16.
- 13. Duman-Scheel M, Weng L, Xin S, Du W. Hedgehog regulates cell growth and proliferation by inducing cyclin D and cyclin E. Nature 2002; 417: 299-304.
- 14. Yoon JW, Kita Y, Frank DJ, et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. J Biol Chem 2002; 277: 5548-55.
- 15. McGarvey TW, Maruta Y, Tomaszewski JE, Linnenbach AJ, Malkowicz SB. PTCH gene mutations in invasive transitional cell carcinoma of the bladder. Oncogene 1998; 17: 1167-72.
- 16. Simoneau AR, Spruck CH 3rd, Gonzalez-Zulueta M, et al. Evidence for two tumor suppressor loci associated with proximal chromosome 9p to q and distal chromosome 9q in bladder cancer and the initial screening for GAS1 and PTC mutations. Cancer Res 1996; 56: 5039-43.
- 17. Xie J, Johnson RL, Zhang X, et al. Mutations of the PATCHED gene in several types of sporadic extracutaneous tumors. Cancer Res 1997; 57: 2369-72.
- 18. Ohgaki K, Minobe K, Kurose K, et al. Two target regions of allelic loss on chromosome 9 in urinary-bladder cancer. Jpn J Cancer Res 1999; 90: 957-64.
- 19. Velickovic M, Delahunt B, McIver B, Grebe SK. Intragenic PTEN/MMAC1 loss of heterozygosity in conventional (clear-cell) renal cell carcinoma is associated with poor patient prognosis. Mod Pathol 2002; 15: 479-85.
- 20. Xu X, Brodie SG, Yang X, et al. Haploid loss of the tumor suppressor Smad4/Dpc4 initiates gastric polyposis and cancer in mice. Oncogene 2000; 19: 1868-74.
- 21. Philipp-Staheli J, Payne SR, Kemp CJ. p27(Kip1): regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res 2001; 264: 148-68.
- 22. Zurawel RH, Allen C, Wechsler-Reya R, Scott MP, Raffel C. Evidence that haploinsufficiency of Ptch leads to medulloblastoma in mice. Genes Chrom Cancer 2000; 28: 77-81.
Liste des figures
Figure 1
Analyse de survie sans récidive par la méthode de Kaplan-Meier, selon la présence de délétions dans les régions 9ptr-p22, 9q22.3, 9q33, 9q34 et 9q34-qtr et pour le chromosome 9 entier.
La présence de délétions de marqueurs microsatellites est associée à un risque plus élevé de récidive dans les régions susceptibles de porter des GST bien qu’on ne retrouve aucune association ailleurs sur le chromosome (9q34-qtr). Le nombre de tumeurs dans chaque catégorie est indiqué au-dessus de la courbe; les cas non informatifs sont exclus de cette analyse. En bleu: courbe de survie avec LOH; en rouge: courbe de survie sans LOH (d’après [6]).
Figure 2
Modèle proposé pour la voie de signalisation ptc/hh chez la drosophile, d’après les données les plus récentes.
A. La voie est inactive lorsque ptc (Patched) se lie à la protéine smo (smoothered) et en inhibe l’activité. Un complexe de protéines cytoplasmiques liées aux microtubules amène le clivage de ci (facteur de transcription cubitus interruptus) en un répresseur transcriptionnel qui exerce son action dans le noyau. B. Voie active: la liaison du ligand hh (hedgedog) à ptc libère smo de l’action répressive de cette dernière, ce qui entraîne la dissociation du complexe cytoplasmique. ci ne subit pas de clivage et agit comme activateur de la transcription de divers gènes dont ptc lui-même. cos-2: protéine costal-2; fu: sérine-thréonine kinase fused; su(fu): suppresseur de fused.
Figure 3
Analyse de l’expression du gène PTC dans des tumeurs vésicales avec perte d’hétérozygotie (LOH) du marqueur PTC, sans LOH détectée sur le chromosome 9, et dans des urothéliums normaux (N).
L’expression du gène a été analysée par la technique de RT-PCR semi-quantitative en présence d’un précurseur radioactif. A. Photographie d’un gel d’électrophorèse montrant la migration des fragments amplifiés pour le gène PTC et pour un gène témoin (glycéraldéhyde-3-phosphate déhydrogénase (GAPDH)). B. Histogramme montrant le rapport de l’intensité des bandes obtenues pour PTC et GAPDH pour les divers échantillons analysés (d’après [7]). On observe une diminution significative de l’expresion du gène dans les tumeurs ayant perdu une copie de PTC.