Résumés
Résumé
Nous explorons dans cette synthèse les forces et les faiblesses de l’écotoxicologie, en nous limitant aux milieux aquatiques. Notre approche consiste à comparer et contraster le comportement des contaminants organiques et inorganiques (métalliques) et à identifier quelques défis pour l’avenir. La prise en charge des contaminants organiques de synthèse se produit le plus souvent par simple diffusion passive au travers d’une membrane cellulaire. Vu la nature lipidique des membranes biologiques, le coefficient de partage octanol-eau (Kow) du contaminant s’avère souvent un bon prédicteur de sa tendance à se bioaccumuler. Par contre, les métaux présents dans le milieu aquatique se trouvent surtout sous des formes hydrophiles et hydratées qui ne peuvent traverser les membranes biologiques par simple diffusion. Leur prise en charge fait alors appel à un transport facilité qui implique des transporteurs protéiques ou canaux transmembranaires. Le coefficient de partage octanol-eau de ces espèces métalliques se révèle inutile comme prédicteur de leur bioaccumulation. Les approches et les modèles prédictifs diffèrent donc grandement entre contaminants métalliques et organiques.
Pour les métaux, deux types de modèles sont couramment employés : des modèles d’équilibre (ex. : le « Modèle du Ligand Biotique » ou BLM) et des modèles cinétiques d’accumulation et d’élimination. Dans les deux cas, les paramètres biologiques des modèles sont considérés comme des « constantes » qui ne sont affectées, ni par la qualité de l’eau ambiante (ex. : pH, dureté), ni par une pré-exposition au métal. Or, il y a maintenant dans la littérature scientifique de plus en plus d’indices que les propriétés clés de la surface épithéliale des organismes aquatiques, qui contrôlent l’accumulation et la toxicité des métaux, ne sont pas constantes, ce qui compromet l’application des modèles dans des cas réels d’exposition chronique sur le terrain. Contrairement aux métaux, l’essentiel du comportement environnemental des composés organiques de synthèse est lié à leur capacité de résister à divers mécanismes de dégradation et à leur biodisponibilité pour les organismes aquatiques. Le modèle de la « fugacité » permet de prédire la distribution de composés organiques entre divers compartiments pour un système considéré à l’équilibre mais de nombreuses contraintes chimiques et biologiques interfèrent avec l’utilisation de ce type de modèle. Les cas des hydrocarbures aromatiques polycycliques (HAP) et des organométaux sont utilisés pour illustrer ces contraintes.
Parmi les tout nouveaux défis de l’écotoxicologie, nous abordons brièvement le développement de la génomique fonctionnelle et de l’approche écosystémique ainsi que la toute nouvelle problématique environnementale posée par les nanoparticules industrielles. L’avenir de l’écotoxicologie aquatique passe nécessairement par : (1) l’obtention de données de terrain et de laboratoire d’excellente qualité; (2) une compréhension approfondie des mécanismes de toxicité aux niveaux moléculaire et cellulaire; (3) le développement de modèles théoriques et empiriques qui intègrent mieux la réalité physiologique et écologique; (4) le développement d’indicateurs écosystémiques capables de fournir une image globale de la qualité d’un environnement aquatique, quelle que soit sa complexité inhérente.
Mots clés:
- écotoxicologie,
- écosystèmes aquatiques,
- métaux traces,
- composés organiques et organométalliques,
- modèle de l’ion libre,
- approche PBT (persistance-bioaccumulation-toxicité),
- modèle BLM,
- modèle de la fugacité,
- nanoparticules
Abstract
In this review we consider the current state of the field of ecotoxicology, with an emphasis on aquatic environments, and explore its strengths and weaknesses. We compare and contrast the environmental behaviour of organic and inorganic contaminants, and identify a number of challenges for the future development of the field. The uptake of synthetic organic contaminants normally occurs by simple passive diffusion across a cell membrane. Given the lipidic and thus hydrophobic nature of biological membranes, the octanol-water partitioning coefficient of an organic contaminant (Kow) is often a good predictor of its tendency to bioaccumulate. In contrast, metals present in the aquatic environment are generally present in hydrated and hydrophilic forms, which cannot cross biological membranes by simple diffusion. Thus their uptake normally occurs by facilitated transport involving membrane carriers or channels. The octanol-water partitioning coefficients of these metallic species thus have no bearing on the relative facility with which they can cross biological membranes. It follows that the modeling approaches will differ greatly between organic and inorganic (metallic) contaminants.
For metals, two types of models are currently popular: equilibrium models (e.g., the “Biotic Ligand Model” or BLM) and kinetic models of metal uptake and elimination. In both cases, the biological parameters of the models are considered as “constants”, which are unaffected by the ambient water quality (e.g., pH; hardness) or by prior exposure to the metal. However, recent research suggests that the key epithelial properties of aquatic organisms that govern metal accumulation and toxicity are not constant, thus compromising the application of the models to real-world cases of chronic exposure to metals. In the case of organic contaminants, ecotoxicological concerns tend to focus on their capacity to resist various degradation mechanisms and on their bioavailability. Fugacity models can be used to predict the distribution of organic molecules among various environmental compartments for systems at equilibrium, but many chemical and biological constraints limit the application of such models. Case studies with polycyclic aromatic hydrocarbons (PAH) and organometallic species are presented to illustrate these constraints.
Finally, among new frontiers and opportunities for ecotoxicology, we briefly consider the development of toxicogenomics, the need to consider the effects of contaminants on trophic interactions in a truly ecosystemic approach, and the challenge posed by nanoparticles of industrial origin. The future of ecotoxicology will necessarily involve: (1) the collection of high quality data in the laboratory and in the field; (2) an improved understanding of the mechanisms of toxicity at the molecular and cellular levels; (3) the development of theoretical and empirical models that better integrate physiological and ecological reality; and (4) the development of ecosystem indicators that can be used to evaluate the quality of aquatic environments, despite their inherent complexity.
Keywords:
- ecotoxicology,
- aquatic ecosystems,
- trace metals,
- organic and organometallic compounds,
- free ion model,
- PBT (persistence-bioaccumulation-toxicity) approach,
- BLM model,
- fugacity model,
- nanoparticles
Corps de l’article
1. Introduction
L’émergence de l’écotoxicologie comme domaine scientifique remonte aux années 1970. On peut retracer son origine à partir de la découverte, pendant la décennie précédente, de la persistance dans l’environnement des pesticides organochlorés et la mise en évidence de leurs effets insidieux sur des espèces non ciblées (CARSON, 1962). Trente années plus tard, plusieurs définitions coexistent pour l’« écotoxicologie » (BOUDOU et RIBEYRE, 1989; BUTLER, 1978; RAMADE, 1979; SHEEHAN et al., 1984; WRIGHT et WELBOURN, 2002). Nous avons retenu la suivante : la science qui cherche à prédire les impacts sur l’écosystème des apports en substances potentiellement toxiques. En adoptant une telle définition, relativement étroite, nous avons consciemment mis l’accent sur la notion des contaminants et leurs effets sur l’écosystème. En effet, certains chercheurs ont élargi la définition de l’écotoxicologie pour y inclure des stresseurs autres que les contaminants (STEINBERG et ADE, 2005), mais pour les fins de cette synthèse, nous nous limiterons à ces derniers.
Selon BUTLER (1978), l’application de l’approche écotoxicologique implique les quatre étapes suivantes : (i) la caractérisation des apports de la substance concernée (quantités, formes physiques et chimiques, et mode d’apport); (ii) l’étude du comportement de la substance dans le milieu récepteur (transport; transformations); (iii) l’évaluation de l’exposition des organismes indigènes à la substance ou à des produits de transformation (espèces cibles; exposition et prise en charge via l’eau ou la nourriture); (iv) l’évaluation de la réponse des organismes individuels, des populations et des communautés à cette exposition. Les étapes (i), (ii) et (iii) s’avèrent relativement simples à franchir; c’est l’étape (iv) qui, dès le début, a été identifiée comme un défi majeur (BUTLER 1978) et qui demeure l’aspect le plus controversé de l’approche (STEINBERG et ADE, 2005). Au final, l’écotoxicologie devrait conduire à une évaluation du risque environnemental pour un écosystème donné. Dans ce cadre général, nous allons explorer les forces et les faiblesses de l’écotoxicologie en nous limitant aux milieux aquatiques. Notre approche consistera à comparer et contraster le comportement des contaminants organiques et inorganiques (métalliques) et à identifier quelques défis pour l’avenir.
2. Comparaison préliminaire des contaminants organiques et inorganiques
Comme déjà mentionné, l’approche écotoxicologique est née en réaction à la découverte de la contamination ubiquiste de l’environnement par des contaminants organiques persistants. Par conséquent, les concepts de base en écotoxicologie se sont développés dans un contexte de composés organiques xénobiotiques. Pour un contaminant de ce type, on se sert normalement de trois critères pour évaluer son danger potentiel : sa persistance dans le milieu récepteur, sa bioaccumulation chez les organismes vivants, et sa toxicité inhérente (PBT). Cette approche s’est avérée utile pour identifier les contaminants problématiques (MACKAY et al., 2001), surtout lorsqu’incorporée dans les protocoles d’évaluation de risques écologiques (US EPA, 1998). Cependant, son application aux composés inorganiques (ex. : les métaux) s’est heurtée à une réalité chimique toute différente. En effet, les contaminants organiques et inorganiques se distinguent nettement sur plusieurs plans (Tableau 1).
Tableau 1
Comparaison – métaux versus contaminants organiques synthétiques (adapté de CAMPBELL et al., 2006).
Comparison of metals and synthetic organic contaminants.
Caractéristique |
Contaminants organiques de synthèse |
Métaux |
|---|---|---|
Origine |
anthropique, sauf pour les hydrocarbures aromatiques polycycliques; |
géogénique (naturellement présents); |
bruit de fond → 0 |
bruit de fond variable |
|
Devenir |
sujets à divers processus de dégradation (hydrolyse, photolyse, biodégradation); |
pas sujets à des processus de dégradation; infiniment persistant, |
demi-vies environnementales utiles; |
demi-vies environnementales sans signification; |
|
composé original → dérivé (changements irréversibles) |
changements de spéciation (réversibles) |
|
bioamplification commune |
bioamplification rare (exception = méthylHg) |
|
Interactions avec des cellules vivantes |
prise en charge se produisant normalement par diffusion à travers une barrière bilipidique; |
prise en charge se produit normalement par transport facilité; |
effets narcotiques communs et toxicité des métabolites |
toxicité spécifique au métal |
|
Fonction biologique |
aucune; mécanismes complexes de défense contre la bioaccumulation des composés toxiques |
souvent biologiquement essentiel; bioaccumulation un phénomène naturel |
Par exemple, si un composé organique synthétique est détecté dans le milieu aquatique, c’est un signe sans équivoque d’une contamination d’origine anthropique, sauf dans le cas particulier des hydrocarbures aromatiques polycycliques (HAP) sur lequel nous reviendrons en détail. Par contre, les métaux sont naturellement présents dans l’environnement, depuis l’origine même de notre planète, et leurs concentrations varient naturellement d’un site à un autre selon la géochimie des roches de surface et les processus d’érosion et lessivage. Cette réalité d’un bruit de fond (« background ») variable fait en sorte que la détection de véritables cas de contamination environnementale soit nettement plus difficile dans le cas de métaux que pour les contaminants organiques (REIMANN et GARRETT, 2005).
Outre cette question de bruit de fond, les deux types de contaminants se distinguent aussi au niveau de leur persistance dans le milieu récepteur. Les contaminants organiques sont sujets à divers processus de dégradation, à la fois abiotiques (hydrolyse; photolyse) et biotiques (biodégradation). La grande majorité de ces processus, normalement irréversibles, impliquent le bris et la formation de liaisons chimiques covalentes menant du composé parent vers la formation de composés filles, généralement rassemblés sous le terme « métabolites ». Les composés organiques se présentent souvent comme des familles de molécules ayant des structures similaires mais de poids moléculaires différents. Les demi-vies des contaminants organiques varient énormément selon la structure moléculaire du contaminant et les conditions environnementales ambiantes (t½ variant entre quelques minutes dans le cas de certaines molécules labiles et plusieurs années pour des produits réfractaires) (SCHWARZENBACH et al., 1993).
D’autre part, dans le cas des métaux, l’élément métallique lui-même est infiniment persistant et la notion de « demi-vie » n’a plus de sens (sauf, bien entendu, dans le cas de radionucléides). Cependant, cette persistance ne s’applique pas nécessairement pour les différentes formes que le métal peut adopter. On fait référence ici à la spéciation de l’élément, c’est-à-dire sa répartition entre différentes formes chimiques ou « espèces » (ex. : différents états d’oxydation; différents complexes). Les transformations entre différentes espèces métalliques impliquent souvent des réactions de complexation (formation ou bris de liaisons coordonnées faibles) ou des réactions d’oxydation/réduction. Contrairement à la situation avec les contaminants organiques, ces réactions sont normalement réversibles, ce qui veut dire que la spéciation d’un métal est largement déterminée par la chimie du milieu dans lequel il se trouve (plutôt que par sa forme initiale, lorsque rejeté dans le milieu récepteur). La notion de spéciation existe aussi pour les contaminants organiques, mais dans ce cas, on parle d’une spéciation moléculaire (liaisons covalentes). La spéciation d’un contaminant organique est alors moins influencée par le milieu dans lequel il se trouve et davantage affectée par sa forme originale au moment où il entre dans le milieu récepteur.
Avant de laisser ces questions de persistance et de spéciation, il faut mentionner le cas particulier des organométaux. Il ne s’agit pas ici de complexes organométalliques où le métal se lie à des ligands via des liaisons coordonnées (faibles), mais plutôt de véritables composés organométalliques où l’atome métallique est lié par une liaison covalente normalement à un atome de carbone ou parfois de soufre. Les composés organométalliques partagent les propriétés du métal capable de former des liaisons ioniques avec les anions en solution (OH‑, Cl‑, CO32‑) et celles du composé organique à la fois liposoluble et biodégradable. Les composés organométalliques de synthèse sont généralement très sensibles à l’oxydation et ne survivent pas en milieu naturel, mais on compte quelques exceptions notoires. Quelques exemples de ces composés seront abordés dans les sections suivantes.
Considérons maintenant l’interaction des contaminants avec les organismes vivants, c’est-à-dire l’exposition. La prise en charge des contaminants organiques de synthèse se produit le plus souvent par simple diffusion passive au travers d’une membrane cellulaire. Vu la nature lipidique des membranes biologiques, le coefficient de partage octanol-eau (Kow) du contaminant s’avère souvent un bon prédicteur de sa tendance à se bioaccumuler. Le passage du contaminant au travers de la membrane cellulaire n’implique pas de transporteur transmembranaire et l’effet biologique du contaminant est souvent caractérisé par la narcose. Il existe toutefois des transporteurs capables d’expulser les toxiques organiques hors de la cellule.
D’autre part, les métaux présents dans le milieu aquatique existent surtout sous formes hydrophiles et hydratées, qui ne peuvent traverser les membranes biologiques par simple diffusion. Leur prise en charge fait alors appel à un transport facilité, normalement passif (c’est-à-dire dans le même sens que le gradient de concentration), qui implique des transporteurs protéiques ou canaux transmembranaires. Le coefficient de partage octanol-eau de ces espèces métalliques se révèle donc inutile comme prédicteur de leur bioaccumulation. Une fois accumulés, les métaux rencontrent une diversité de ligands dans le milieu intracellulaire dont certains conduisent à une détoxication efficace. Il en résulte que la concentration intracellulaire totale du métal comprend à la fois des formes détoxiquées et des formes bioactives et s’avère peu utile comme prédicteur de toxicité, contrairement à ce qui prévaut pour les contaminants organiques souvent bioaccumulés dans les vacuoles lipidiques et la membrane cellulaire elle-même. Une complication supplémentaire apparaît lorsque les métabolites demeurent suffisamment liposolubles pour être bioaccumulés et exercer leurs effets toxiques sur la cellule.
Toujours au niveau des interactions contaminants-organismes, il convient de se rappeler ici l’ubiquité des métaux. Les organismes vivants ont effectivement « appris » à vivre en présence de métaux, à en profiter même, en les incorporant dans leur métabolisme (ex. : comme coenzymes ou comme cofacteurs). Plusieurs métaux traces sont biologiquement essentiels (ex. : Co, Cu, Ni, Zn). Leur présence dans le milieu ambiant est une condition nécessaire à la vie et leur bioaccumulation est un phénomène tout à fait naturel – il n’en sera pas ainsi pour les contaminants organiques.
Ayant identifié des différences importantes entre les contaminants organiques et inorganiques, nous traiterons chaque classe de contaminants séparément dans les prochaines sections. Pour chaque type de contaminants, nous ferons un bilan des connaissances actuelles en écotoxicologie (en nous limitant aux milieux aquatiques) et nous ferons ressortir quelques défis pour l’avenir, notamment à propos de nanoparticules de synthèse.
3. Courants modernes (écotoxicologie des métaux)
3.1 Sources de métaux
Pour prédire le comportement d’un métal dans l’environnement et ses effets sur l’écosystème, il importe peu de savoir si le métal provient d’une source naturelle (ex. : volcans; bouches hydrothermales) ou d’une source anthropique. Cependant, en pratique, lorsqu’on cherche à gérer les risques pour l’écosystème, il s’avère très important de pouvoir faire cette distinction; le succès des mesures de mitigation choisies dépendra de l’importance relative des sources anthropiques (c’est-à-dire celles qui sont contrôlables) par rapport à celles qui sont naturelles. NRIAGU (1989) a été le premier à tenter de quantifier l’importance relative des sources naturelles et anthropiques à l’échelle globale, mais il a été confronté à des lacunes importantes dans les données de base. Le sujet demeure important et plusieurs autres chercheurs ont repris les calculs de NRIAGU, parfois à l’échelle globale (GARRETT, 2000; PACYNA et al., 1995; PACYNA et al., 2006), mais souvent pour des régions spécifiques ou pour certains métaux individuels (OLENDRZYNSKI et al., 1996; RICHARDSON et al., 2003). Ces analyses démontrent clairement que les activités anthropiques ont perturbé les cycles géochimiques de plusieurs métaux, dont Ag, Cd, Cu, Hg, Ni, Pb et Zn.
3.2 Devenir des métaux dans le milieu récepteur; modèles de spéciation
Tel que mentionné dans l’introduction, la prédiction du comportement des contaminants dans le milieu récepteur fait partie intégrante de l’approche écotoxicologique (évaluation de l’exposition des organismes vivant dans l’écosystème). Nous avons besoin de modèles qui permettent de prédire les concentrations du contaminant dans divers compartiments environnementaux, et ceci en fonction des apports de ce contaminant. De tels modèles ont été développés et testés pour des contaminants organiques hydrophobes (SCHWARZENBACH et al., 1993) et, grâce à des relations de type QSAR (relation quantitative structure-activité), on peut même faire des prédictions fiables pour de nouveaux composés. Pour les métaux, cependant, de telles prédictions demeurent problématiques (PAQUIN et al., 2003; US EPA, 2007) et par conséquent nous ne pouvons pas simuler avec confiance les effets d’une éventuelle réduction des apports anthropiques d’un métal donné, par exemple. Cette lacune constitue une barrière importante à l’application pratique de l’approche écotoxicologique aux métaux.
Au niveau moléculaire, le devenir des métaux cationiques dans le milieu aquatique et leur éventuelle biodisponibilité sont en grande partie contrôlés par leur spéciation, laquelle est déterminée par les conditions physico-chimiques ambiantes (ex. : pH, dureté, salinité, concentration en matière organique dissoute (MOD)). Compte tenu de cette influence marquée de la spéciation des métaux dissous, il importe de pouvoir la prédire. Il existe plusieurs modèles d’équilibre chimique qui permettent de calculer la répartition d’un métal donné entre différentes formes chimiques (ex. : l’ion métallique libre, Mz+, ainsi que ses complexes). Pour les complexes inorganiques (ex. : hydroxo-, chloro-, sulfato-, carbonato-complexes), et pour des solutions relativement diluées, ces modèles performent bien et fournissent des prédictions précises et utiles (TURNER, 1995).
Cependant, les eaux naturelles contiennent aussi des quantités appréciables mais variables d’acides fulviques et humiques. Ces acides organiques naturels ont la capacité de complexer les métaux cationiques, mais cette complexation ne se prête pas à une modélisation simple (DUDAL et GÉRARD, 2004). En effet, les acides fulviques et humiques ne se comportent pas comme des ligands organiques simples, mais plutôt comme des polyélectrolytes multifonctionnels. Deux modèles ont été développés pour tenir compte de ces propriétés : le « Windermere Humic Aqueous Model » or WHAM (TIPPING, 1998), et le « Nonideal Competitive Adsorption-Donnan » ou NICA-Donnan (MILNE et al., 2001; MILNE et al., 2003). Une fois calibrés, ces modèles réussissent bien à reproduire les résultats obtenus en laboratoire lors de titrages d’acides fulviques ou humiques isolés (TIPPING, 2002). Une question évidente se pose cependant : ces modèles performent-ils aussi bien lorsqu’appliqués à des eaux naturelles? Une seule étude sérieuse a été réalisée pour vérifier cette question (SIGG et al., 2006; UNSWORTH et al., 2006). Ces chercheurs ont d’abord déterminé la spéciation de plusieurs métaux (Cd, Cu, Ni, Pb, Zn) dans différentes eaux naturelles et ensuite comparé ces résultats aux prédictions des modèles WHAM et NICA-Donnan. Dans le cas de l’ion métallique libre (Mz+), l’espèce clé qui est reconnue comme le meilleur prédicteur de la biodisponibilité d’un cation métallique dissous (CAMPBELL, 1995), il y avait des divergences énormes entre les valeurs mesurées expérimentalement et les prédictions des modèles (ex. : les valeurs mesurées pour le Cu2+ étaient jusqu’à 105 plus élevées que les concentrations prédites!). Il s’agit là d’un résultat inattendu et très décevant. Vu l’importance toxicologique de l’ion métallique libre, il y a un besoin urgent d’approfondir et d’expliquer ce résultat. Pour le moment, il est clair que la capacité des modèles actuels à reproduire la spéciation des métaux cationiques dans les eaux naturelles reste à démontrer.
3.3 Effets des métaux (organismes individuels)
3.3.1 Métaux dissous
Afin de prédire les effets des métaux sur les organismes aquatiques, il faut d’abord déterminer comment les métaux interagissent avec les organismes aquatiques (CAMPBELL, 1995; CAMPBELL et COUILLARD, 2004). Trois mécanismes de prise en charge ont été identifiés (Figure 1) : (i) transport passif, par simple diffusion, d’un complexe métallique neutre (MLn0) et lipophile (ex. : HgCl20); (ii) transport facilité du cation (Mz+), impliquant soit un transporteur transmembranaire protéique, soit un canal transmembranaire; et (iii) transport facilité d’un complexe métallique anionique (MZ‑), impliquant un transporteur d’anions relativement peu sélectif (transport « accidentel » de l’anion + le métal qui y est associé). Normalement, c’est la voie (ii) qui prédomine, et dans de tels cas, c’est le « Modèle du ligand biotique » ou « Biotic Ligand Model (BLM) » qui s’applique. Selon ce modèle (PAQUIN et al., 2002), le métal se lie initialement à des sites de liaison « X-membrane » sur la membrane cellulaire, ces sites étant considérés comme des ligands en solution avec leurs propres constantes de stabilité. À condition que l’équilibre s’établisse rapidement entre le métal en solution et la surface biologique (« rapidement » signifie plus vite que l’expression de la réponse toxique), la réponse biologique sera fonction du nombre de sites « sensibles » occupés par le métal M, i.e. la concentration <M‑X-membrane>, où les accolades < > désignent la concentration d’une espèce formée à la surface cellulaire.
Figure 1
Prise en charge de métaux par des organismes aquatiques – mécanismes généraux : (i) diffusion passive de complexes métalliques lipophiles; (ii) transport facilité de cations; (iii) transport accidentel de cations liés à des anions.
Metal uptake by aquatic organisms – general mechanisms: (i) passive diffusion of lipophilic metal complexes; (ii) facilitated cation transport; (iii) accidental transport of cations bound to anions.

La réaction du métal à la surface cellulaire implique la formation d’un complexe métallique : M(L) + X membrane ↔ M‑X membrane + L, où M = cation métallique, L = ligand associé au métal en solution, X = site de liaison intégré dans la membrane épithéliale (les charges électriques sur les diverses espèces ne sont pas indiquées). Si le site X-membrane correspond à un site physiologiquement actif présent à la surface cellulaire, sa réaction avec le métal pourra induire une réponse biologique directe. D’autre part, si X-membrane correspond plutôt à un site de transport qui permet au métal de traverser la membrane cellulaire et entrer dans la cellule, alors sa réaction avec le métal ne constituera qu’une étape préalable au transport membranaire (et la réaction du métal avec les sites physiologiquement sensibles se produira dans le milieu intracellulaire, suite au transport). Dans une variante de ce scénario, X‑membrane pourrait correspondre à un site de transport normalement emprunté par un oligo-élément nutritif essentiel; dans ce cas, sa réaction avec le métal « M » inhiberait le transport du métal essentiel et y induirait une déficience. Dans les trois cas, la réponse biologique devrait être proportionnelle à la concentration du complexe de surface <M‑X‑membrane>.
Cette description simplifiée de l’interaction des métaux avec les organismes vivants démontre le lien entre la spéciation du métal et sa biodisponibilité et fait ressortir l’importance de la concentration de l’ion métallique libre comme prédicteur de cette biodisponibilité. Selon ce modèle, la complexation du métal en solution devrait mener à une diminution de sa biodisponibilité et de telles diminutions sont effectivement observées dans la grande majorité des cas (CAMPBELL, 1995). Cependant, pour prédire les effets biologiques d’un métal, la concentration de l’ion métallique libre ne suffit pas à elle seule. Il faut aussi tenir compte de la dureté de l’eau (compétition entre Mz+ et Ca2+ ou Mg2+ pour le site X‑membrane) et du pH (compétition entre l’ion Mz+ et H+ pour ce même site). L’ensemble de ces réactions se trouve intégré dans le BLM, qui a été largement adopté par les agences responsables de la protection de l’environnement aux États-Unis, au Canada, en Europe ainsi qu’en Australie (NIYOGI et WOOD, 2004).
Le développement et l’acceptation du BLM représentent un succès indéniable, dans le sens que, pour une première fois, les notions de spéciation et de biodisponibilité sont prises en compte par les acteurs responsables de la protection de l’environnement. Cependant, le modèle a été développé et calibré avec les résultats de tests de toxicité aiguë (concentrations métalliques élevées, expositions courtes), et son application à des cas de toxicité chronique soulève plusieurs questions, notamment en ce qui concerne la possibilité d’acclimatation. Le BLM est essentiellement un modèle d’équilibres chimiques qui prédit la répartition du métal d’intérêt entre divers ligands (ceux qui sont libres en solution et également le « ligand biotique » qui se trouve à la surface cellulaire, que nous avons désigné « X‑membrane »). Selon ce modèle, les propriétés du « ligand biotique » sont considérées comme des paramètres biologiques invariables qui ne sont pas affectés par la qualité de l’eau ambiante (pH, dureté, [MOD]) ou par une pré-exposition au métal. Or, il y a maintenant dans la littérature scientifique de plus en plus d’indices que les propriétés clés de la surface épithéliale (ex. : sa capacité à lier le métal; son affinité pour le métal) ne sont pas constantes. Par exemple, dans une étude en laboratoire sur la truite arc-en-ciel, KAMUNDE et al. (2002) ont démontré que la pré-exposition des poissons au cuivre pendant 28 j avait pour effet de perturber les interactions Cu‑branchie : l’affinité de la branchie pour le cuivre a changé au cours de la pré-exposition (moins de sites de haute affinité, davantage de sites de faible affinité) et la prise en charge du cuivre, mesurée avec le 64Cu pendant de courtes périodes d’exposition, a diminué. De même, en comparant des poissons récoltés dans un lac propre à ceux provenant d’un lac contaminé en métaux, KLINCK et al. (2007) ont constaté d’importantes différences au niveau de l’affinité des branchies pour les métaux et la prise en charge des métaux, suggérant que les poissons vivant dans le lac contaminé s’étaient acclimatés pendant leur exposition chronique. Toute application du BLM pour prédire la toxicité chronique des métaux devra tenir compte de ces observations, par exemple, en incorporant une gamme de propriétés pour le ligand biotique plutôt qu’une seule combinaison de KML et de « L ».
3.3.2 Métaux présents dans la nourriture
Outre la question d’acclimatation, il faut aussi se rappeler que le BLM ne considère que l’exposition via l’eau ambiante, avec l’hypothèse implicite que les métaux apportés par ingestion ne provoquent pas d’effets. Il s’agit d’une hypothèse simplificatrice qui a peut-être sa raison d’être lorsque les concentrations métalliques aqueuses sont très élevées (toxicité aiguë), mais qui manque de fondement scientifique pour des conditions d’exposition chronique (FISHER et HOOK, 2002; HOOK et FISHER, 2001; MEYER et al., 2005). Heureusement, en parallèle au développement du BLM, d’autres chercheurs se sont penchés sur le transfert trophique des métaux, c’est-à-dire sur le rôle de la nourriture comme vecteur d’exposition aux métaux. Dans sa forme la plus simple (LUOMA et RAINBOW, 2005), cette approche biodynamique assimile l’organisme à un seul compartiment et considère deux vecteurs d’accumulation et une voie d’élimination (Figure 2). La concentration du métal chez l’organisme à l’état stationnaire, [M]es, est donnée par l’équation suivante
où ku = constante de vitesse pour la prise en charge à partir de l’eau (L-1 g‑1 j‑1); ke = constante de vitesse pour l’efflux (j‑1), kg = constante de croissance (j‑1); [M]eau = concentration du métal dissous (mol·L‑1); [M]nourriture = concentration du métal dans la nourriture (mol·g‑1), EA = efficacité d’assimilation (0 → 1,0), et TI = taux d’ingestion (g nourriture·g‑1 organisme). D’habitude, les valeurs de ku, ke, EA et TI sont déterminées dans des expériences de courte durée et les valeurs de kg viennent d’observations sur le terrain.
Figure 2
Modélisation biodynamique : représentation simplifiée (modèle à un compartiment) de l’accumulation et de l’élimination d’un contaminant par un organisme aquatique (LUOMA et RAINBOW 2005). Les paramètres du modèle sont expliqués dans le texte.
Biodynamic modeling: simplified representation (one compartment model) of the accumulation and elimination of a contaminant by an aquatic organism (LUOMA and RAINBOW, 2005). The model parameters are explained in the text.

Dans leur récent article de synthèse, LUOMA et RAINBOW (2005) ont identifié 15 publications où ils ont pu comparer les prédictions du modèle avec les observations faites sur le terrain. Pour l’ensemble de 7 métaux et 14 espèces animales, les concentrations pour le corps entier couvraient une gamme de 107, témoignant de la grande variabilité d’une espèce à une autre et d’un métal à un autre pour une espèce donnée. Après transformation logarithmique des données et leur mise en commun, le coefficient de détermination globale (r2) entre prédictions et observations étaient de 0,98; dans la plupart des cas, l’écart entre l’observation et la prédiction était inférieur à un facteur de deux.
3.3.3 Intégration des modèles (BLM et biodynamique)
Les deux approches de modélisation décrites ci-haut apparaissent complémentaires, l’approche thermodynamique (BLM) s’appliquant davantage aux cas de toxicité aiguë et l’approche cinétique étant plus appropriée pour les cas de toxicité chronique. Cependant, pour vraiment profiter de l’approche cinétique, il faudrait pouvoir la raffiner pour prédire l’accumulation des métaux, non seulement dans l’organisme entier, mais également dans des organes cibles (la toxicité des métaux étant généralement spécifique à un organe). Nous avons récemment testé un tel modèle biodynamique d’accumulation du cadmium chez la perchaude juvénile (Perca flavescens : branchies, intestin et foie). Le modèle a été calibré avec des données cinétiques obtenues dans le cadre d’expériences antérieures menées sur le terrain (transplantation d’un milieu propre à un milieu contaminé en Cd; manipulation du régime alimentaire – KRAEMER et al. (2008)). Les simulations pour le Cd hépatique se conformaient bien aux valeurs observées, mais pour les branchies et l’intestin, les valeurs prédites à la fin de la période de 70 j de transplantation étaient environ trois fois supérieures à celles observées, ce qui suggère que ces derniers organes puissent changer leur capacité de prendre en charge ou d’éliminer le Cd lorsque les concentrations accumulées dépassent un certain seuil.
Le modèle a également été utilisé pour prédire les concentrations en Cd à l’état stationnaire dans les branchies, l’intestin et le foie de perchaudes indigènes vivant dans des lacs situés le long d’un gradient en Cd. L’accord entre les simulations et les concentrations en Cd observées était acceptable dans des lacs ayant des concentrations en Cd faibles ou modérées, mais dans les lacs les plus contaminés en Cd ([Cd] > 1,5 nM), le modèle a de nouveau surestimé l’accumulation, surtout dans les branchies et l’intestin. Ces résultats suggèrent que les modèles cinétiques de bioaccumulation s’appliquent mieux à certains organes qu’à d’autres, et ils laissent entrevoir aussi que les constantes de vitesse pour l’accumulation des métaux dans un organe ou pour leur élimination ne soient pas de véritables « constantes », mais qu’elles puissent varier en fonction du régime d’exposition. Cette situation ressemble à celle décrite ci-haut pour le BLM (variabilité des propriétés du ligand biotique) et illustre bien le défi d’intégrer les phénomènes d’acclimatation et d’homéostasie dans les modèles de toxicité pour les métaux.
3.3.4 Répartition subcellulaire des métaux
Outre le défi de pouvoir prédire l’accumulation d’un métal donné dans les divers organes d’un organisme cible, il serait également intéressant de pouvoir tenir compte de sa répartition subcellulaire dans l’organe en question. Plusieurs organismes réussissent à détoxiquer les métaux en les séquestrant sous une forme inactive (MASON et JENKINS, 1995). Dans de tels cas, il serait hasardeux de vouloir prédire la toxicité du métal en fonction de sa concentration totale dans un organe donné. Ce genre de raisonnement explique pourquoi de plus en plus de chercheurs s’intéressent à la répartition subcellulaire des métaux (c.-à-d. leur « spéciation intracellulaire ») (CAMPBELL et al., 2005; VIJVER et al., 2004; WANG et RAINBOW, 2006).
Selon ce modèle conceptuel de toxicité/détoxication (CAMPBELL et COUILLARD, 2004; MASON et JENKINS, 1995), la détermination de la répartition intracellulaire des métaux pourrait renseigner sur l’état physiologique d’un organisme. Pour tester ce modèle, il faut pouvoir différencier expérimentalement les métaux détoxiqués (ex. : associés à des granules, à des vacuoles digestives, à la métallothionéine) des métaux bioactifs. Si on se limite au cytosol, c’est-à-dire aux ligands intracellulaires dissous (acides aminés, peptides, métallothionéine, enzymes, etc.), on peut homogénéiser le tissu, isoler le cytosol par ultracentrifugation, et ensuite séparer les complexes cytosoliques par chromatographie d’exclusion (MASON et al., 1990). Dans ce genre d’approche, on associe les ligands de poids moléculaire élevé à des enzymes, une fraction qui, normalement, ne devrait pas contenir des métaux non essentiels. En suivant de près leur contenu en métaux, on peut en principe déceler l’initiation du « débordement cellulaire » ou « spillover ». Pour considérer à la fois le cytosol, les organelles ainsi que les granules / lysosomes, on peut faire appel à des techniques de centrifugation différentielle (GIGUÈRE et al., 2006; WALLACE et al., 2003) et les coupler à une étape finale de chromatographie d’exclusion, appliquée au cytosol.
Nous avons récemment testé ce modèle de « débordement cellulaire » sur le terrain (CAMPBELL et al., 2008; GIGUÈRE et al., 2006). Des perchaudes (P. flavescens) ont été récoltées dans des lacs situés le long d’un gradient de contamination en Cd et en Ni, et nous avons déterminé les concentrations totales de ces métaux dans les eaux de lacs (comme mesure de l’exposition) et dans le foie des poissons (comme mesure de l’accumulation totale). À l’aide d’un protocole de centrifugation différentielle adapté à la perchaude, nous avons également étudié la répartition des métaux hépatiques parmi des fractions potentiellement sensibles aux métaux (protéines, organelles) et les fractions représentant les métaux détoxiqués (métallothionéine; granules) – voir figure 3. Le Cd hépatique se trouvait majoritairement dans le cytosol, lié à des protéines thermostables (métallothionéines), mais il y en avait aussi dans les fractions potentiellement sensibles aux métaux (protéines thermosensibles et organelles); quant au Ni hépatique, il était surtout associé aux protéines thermosensibles. Mettre en évidence le « débordement » de ces métaux, c’est-à-dire détecter leur présence dans les fractions où normalement ils ne devraient pas se trouver, n’est peut-être pas surprenant. Cependant, pour ces poissons exposés chroniquement à des métaux, il n’y avait pas de concentration seuil au-dessous de laquelle ces fractions sensibles aux métaux étaient complètement protégées. En d’autres termes, la détoxication des métaux était clairement incomplète, les poissons étant sujets à des stress induits par les métaux même dans les lacs faiblement contaminés (GRAVEL et al., 2005; LEVESQUE et al., 2003).
Figure 3
Concentrations en Cd hépatique dans diverses fractions subcellulaires chez la perchaude jaune, Perca flavescens: (a) protéines thermosensibles, (b) organelles et (c) protéines thermostables. Les résultats sont présentés en fonction des concentrations hépatique totales en Cd dans les poissons récoltés dans des lacs le long d’un gradient de concentrations métalliques (CAMPBELL et al., 2008). Les données proviennent d’études spatiales en 2001 (cercles) et en 2003 (triangles). Dans le graphique (b), les cercles solides = lysosomes + microsomes; les cercles ouvertes = mitochondries. Chaque point correspond à la valeur moyenne pour les poissons récoltés à une seule station d’échantillonnage dans un lac donné. Figure adaptée de CAMPBELL et al. (2008), avec la permission de la maison de publication Taylor et Francis.
Concentrations of hepatic Cd in various subcellular fractions in yellow perch, Perca flavescens: (a) heat-denaturable proteins, (b) organelles and (c) heat-stable proteins. The results are presented as a function of total hepatic Cd in juvenile perch sampled along a metal concentration gradient (CAMPBELL et al., 2008). Data come from spatial studies in 2001 (circles) and 2003 (triangles). In panel (b), solid circles = lysosomes and microsomes, open circles = mitochondria. Each point represents data for individual fish collected at a single sampling station. Figure adapted from CAMPBELL et al. (2008), with the permission of the publisher, Taylor and Francis.
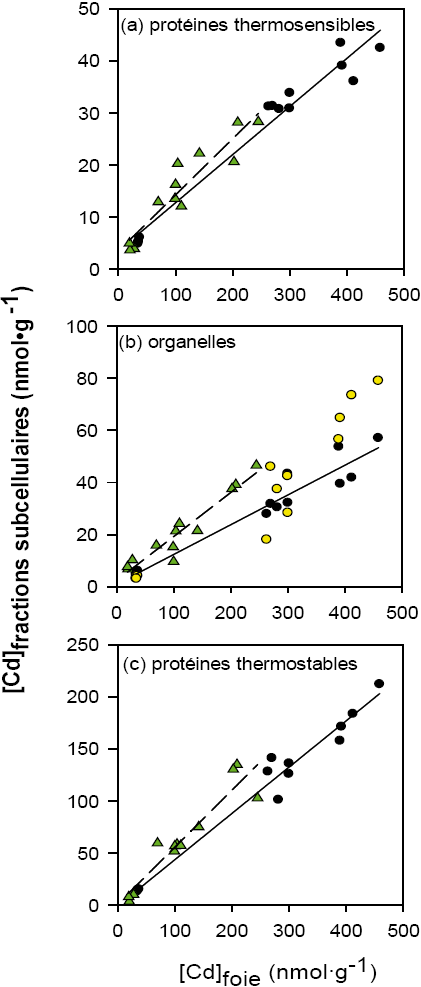
Il s’agit ici d’un résultat inattendu, apparemment sans précédent, qui mine la crédibilité du modèle classique de toxicologie des métaux (MASON et JENKINS, 1995). Ce modèle postule l’existence d’un seuil d’accumulation au-dessous duquel l’organisme cible réussit à détoxiquer complètement le métal. Nos résultats suggèrent au contraire que les organismes chroniquement exposés aux métaux puissent accepter un compromis entre deux options, c’est-à-dire détoxiquer le métal partiellement mais permettre à une fraction du métal de se lier à des sites inappropriés (tout en économisant sur le coût métabolique de la détoxication).
3.3.5 Génétique fonctionnelle
Avant de laisser le sujet des effets des métaux sur les organismes individuels, il importe de mentionner brièvement l’influence grandissante de la biologie moléculaire dans ce domaine (ANKLEY et al., 2006; HOGSTRAND et al., 2002; PELLETIER et al., 2004). Plusieurs laboratoires ont reconnu le potentiel qu’offre la génomique, notamment pour élucider les mécanismes de toxicité et de détoxication (identifier le mode d’action de la substance toxique et reconnaître les exemples de réponse qui caractérisent certaines familles de stresseurs), mais aussi pour déterminer si les populations naturelles sont affectées par leur exposition (ponctuelle ou chronique). On parle ici de la « toxicogénomique », soit l’application de la génomique à la toxicologie qui comprend l’étude de l’expression génétique (ARN messager) et de la transcription de l’ARN messager (le protéome, le métabolome).
Les premiers essais avec les métaux, réalisés en laboratoire avec des espèces aquatiques typiques (ex. : poisson zèbre, truite arc-en-ciel, daphnies), datent d’il y a à peine cinq ans. Malheureusement, les chercheurs ont employé des concentrations métalliques complètement irréalistes, probablement parce qu’ils voulaient être certains de détecter une réponse. Les expositions correspondaient à des conditions qui ne se produiraient jamais dans le milieu récepteur réel. Par conséquent, les résultats demeurent difficiles à interpréter dans un contexte environnemental. Maintenant assurés de pouvoir détecter une réponse, quelques chercheurs ont repris ces premières expériences mais avec des concentrations métalliques nettement plus faibles (ex. : GONZALEZ et al., 2006; PIERRON et al., 2007). Les résultats sont prometteurs, mais il est encore trop tôt pour reconnaître des modes d’action pour un métal donné qui se manifestent systématiquement chez plusieurs espèces. Si on réussit à identifier de tels modes, ceux-ci pourraient être utilisés pour détecter sans équivoque un stress métallique chez les organismes sauvages exposés dans leur milieu. Il s’agit là du rêve de plusieurs écotoxicologues de pouvoir diagnostiquer l’état de santé des organismes sauvages.
3.4 Effets des métaux (écosystème)
Jusqu’ici nous avons mis l’accent sur les effets des métaux sur les organismes individuels, en discutant de leur interaction à la surface cellulaire, de leur transport au travers de la membrane cellulaire et, finalement, de leur devenir au niveau subcellulaire (détoxication). En choisissant cette approche classique, nous avons forcément considéré les effets directs des métaux sur divers organismes cibles. Cependant, il faut se rappeler que l’écotoxicologie vise à prédire les effets des contaminants non seulement sur les organismes individuels, mais aussi au niveau des populations et des communautés. En effet, il arrive parfois que les effets des métaux soient indirects, c’est-à-dire transmis via la chaîne trophique.
La contamination par les métaux peut perturber les réseaux trophiques en modifiant la composition taxonomique, la structure et la taille des communautés de macroinvertébrés benthiques. Ces derniers jouent un rôle important dans le milieu aquatique en servant, entre autres, de proies pour les populations de poissons benthivores. Nous avons récemment mis en évidence de tels effets indirects sur les populations de perchaude (Perca flavescens) vivant dans des lacs situés sur le Bouclier canadien le long d’un gradient de contamination métallique (Cd, Cu, Zn) (CAMPBELL et al., 2003; RASMUSSEN et al., 2008). La perchaude étant relativement tolérante aux concentrations élevées en métaux ainsi qu’aux conditions modérément acides, elle domine souvent les communautés piscicoles dans les lacs nord-américains affectés par l’exploitation minière. Au cours de son développement normal, en grandissant dans un lac propre, la perchaude ajuste successivement son régime alimentaire : planctivore → benthivore → omnivore.
Des perchaudes ainsi que des macroinvertébrés benthiques ont été prélevés dans chaque lac. Dans les lacs les plus contaminés, la densité de plusieurs taxons de macroinvertébrés benthiques était significativement plus basse et la diversité taxinomique était significativement plus faible que dans les lacs moins contaminés. Les contenus stomacaux des perchaudes reflétaient cette même tendance, étant moins diversifiés chez les poissons provenant de lacs contaminés que chez ceux prélevés dans les autres lacs étudiés. Les chironomes et (ou) le zooplancton étaient davantage présents dans les contenus stomacaux de perchaudes vivant dans les lacs les plus contaminés. De plus, dans ces lacs contaminés, la taille moyenne des proies était plus faible que dans les lacs moins contaminés. Enfin, dans les lacs contaminés, la taille des proies trouvées dans les contenus stomacaux n’augmentait pas en fonction de l’âge des perchaudes. On a aussi observé que pour des spécimens du même âge, les poissons provenant des lacs à contamination de moyenne à faible pesaient significativement plus que ceux trouvés dans des lacs à forte contamination. Cette réduction de la croissance a été attribuée aux coûts accrus de la recherche de nourriture dans le réseau simplifié de proies qui caractérise les systèmes contaminés par les métaux (RASMUSSEN et al., 2008).
Ces observations sont importantes puisqu’elles suggèrent que des niveaux trophiques élevés (en l’occurrence, les poissons) puissent être affectés par les métaux même si les concentrations métalliques ambiantes ne sont pas assez élevées pour y nuire directement. Il s’agit d’un des rares exemples de l’intégration de l’écologie dans l’approche écotoxicologique (STEINBERG et ADE, 2005).
4. Courants modernes (écotoxicologie – composés organiques et organométaux)
4.1 Sources des composés organiques
La très vaste majorité des molécules organiques présentant un intérêt écotoxicologique ont une origine anthropique, sauf pour l’importante famille des hydrocarbures aromatiques polycycliques (HAP) provenant de la combustion incomplète de toute matière organique fossile ou fraichement synthétisée par l’activité biologique. Dans ce cas particulier, la difficulté à différencier les apports naturels (feux de forêts et volcanisme) des apports anthropiques (combustion des carburants fossiles et du bois) est la même que celle rencontrée pour les métaux. Dans tous les autres cas, la présence d’une molécule spécifique (comme un herbicide ou un insecticide) ou de toute une famille de composés apparentés (comme les biphényles polychlorés : BPC) est obligatoirement reliée à une origine anthropique, même si la source présumée se trouve à des milliers de km du point d’impact. Les meilleurs exemples de la dispersion planétaire de molécules de synthèse restent les organochlorés comme le dichlorodiphényltrichloroéthane (DDT) et les biphényles polychlorés (BPC) (WANIA et MACKAY, 1993). Produits à l’échelle industrielle après la seconde guerre mondiale, ces composés ont trouvé de multiples usages industriels et agricoles et ont peu à peu envahi les sols, les cours d’eau, les estuaires et les sédiments marins. Interdits de fabrication et d’utilisation dans la plupart des pays industrialisés depuis le début des années 1970, on croyait à leur disparition en quelques années mais on sous-estimait grandement leur persistance dans l’environnement. Il n’est pas rare de retrouver encore aujourd’hui des centaines de mg/kg de ces composés dans les tissus des mammifères marins qui fréquentent les zones côtières de l’Amérique du Nord et de l’Europe (HANSEN et al., 2004; ROSS et al., 2000). À ceux-ci se sont ajoutés plus récemment les polybromodiphényléthers (PBDE), une toute nouvelle famille de 209 congénères ayant des propriétés ignifugeantes et comportant une structure polycyclique à laquelle sont attachés des atomes de brome en nombre variable (Figure 4) et pour lesquels nous n’avons que de maigres informations sur la toxicité aquatique et le potentiel de dispersion dans l’environnement (HALE et al., 2003). Quelques travaux de modélisation permettent de comparer le comportement des BPC et des PBDE dans les chaines trophiques (VELTMAN et al., 2005). Le marché mondial des polybromés était d’environ 120 000 tonnes en 2001 et continue à croître.
Figure 4
Structure générale des polybromo-diphényléthers (PBDE).
General structure of polybromo-diphenylethers (PBDE).
Notons enfin que l’agriculture intensive et l’élevage sont des sources permanentes de molécules de synthèse souvent considérées à tort comme rapidement biodégradables et sans effet à long terme sur l’environnement. Ainsi, certains herbicides de la famille des chloracétanilides comme le métolachlore ont un temps de demi-vie cinq à six fois plus élevé dans un sédiment anoxique que dans la colonne d’eau (MERSIE et al., 2004) et les insecticides organophosphorés comme le diazinon et le chlorpyrifos peuvent persister plusieurs mois dans les eaux marines côtières (BONDARENKO et al., 2004) comparativement à quelques jours dans les eaux douces d’une rivière.
Même en possession de la panoplie toujours plus sophistiquée des équipements analytiques de haute technologie, le défi le plus important du chimiste analyste en environnement reste la détection et la quantification des milliers de molécules organiques de synthèse qui atteignent les milieux aquatiques et sédimentaires chaque année. Le danger intrinsèque d’une molécule de synthèse est souvent bien connu par les études en laboratoire mais le risque environnemental qui y est associé est intimement lié à sa présence dans l’environnement et à sa disponibilité pour les organismes vivants, de la bactérie jusqu’à l’humain.
4.2 Modéliser le devenir des composés organiques dans le milieu récepteur
Contrairement aux métaux qui sont pour ainsi dire éternellement recyclables, tous les composés organiques libérés dans notre environnement planétaire sont sensibles à des réactions de dégradation, que ce soit par : (i) la photolyse sous l’action des rayons du soleil; (ii) l’hydrolyse sous l’action des molécules d’eau; (iii) la combustion sous l’action directe de l’oxygène ou encore (iv) la biodégradation par l’action des organismes vivants. L’action individuelle ou la combinaison de tous ces processus conduit invariablement à la production de CO2 qui pourra aussitôt reprendre du service et redevenir une molécule organique par la photosynthèse ou le produit d’un procédé catalytique inventé par la chimie industrielle. L’essentiel du comportement environnemental des composés organiques de synthèse est lié à leur capacité à résister aux divers mécanismes de dégradation énoncés plus haut et à leur biodisponibilité pour les organismes qui vivent dans cet environnement.
On peut tenter de prévoir le comportement et le devenir d’un composé organique donné en connaissant bien ses propriétés environnementales comme sa solubilité dans l’eau, sa volatilité en fonction de la température de l’eau et de l’air et sa propension à s’associer aux particules minérales ou organiques qui forment les sols et les sédiments. En fait, le problème peut se résumer ainsi : comment parvenir à quantifier la tendance qu’aura un composé organique à se distribuer entre les trois phases de l’environnement : l’air, l’eau et le géosorbant (sol ou sédiment)? Ce problème peut se résoudre à partir des propriétés physico-chimiques du composé étudié et par l’application rigoureuse des principes de base de la thermodynamique et de la cinétique chimique. On peut donc modéliser le comportement d’une molécule organique même dans un environnement complexe. L’une des approches les mieux connues est le modèle de la « fugacité » introduit dans un contexte environnemental par le professeur Don Mackay de l’Université de Toronto à la fin des années 1970 (MACKAY, 1979; MACKAY et PATERSON, 1982) et ensuite largement développé dans de nombreux articles et livres spécialisés (CAHILL et al., 2003; MACKAY, 1991; MACKAY et FRASER, 2000).
La fugacité peut être illustrée comme la « tendance qu’aura un composé à fuir un compartiment environnemental pour se réfugier dans un autre ». Elle détermine la tendance d’une substance à préférer une phase (gaz, liquide ou solide) par rapport à une autre. Pour une température et une pression données, une substance organique aura une fugacité qui sera différente pour chacune des phases, prises indépendamment l’une de l’autre et avant équilibrage du système. La phase avec la plus faible fugacité sera la plus favorisée. La fugacité n’est pas une propriété physique d’une substance mais une propriété calculée que l’on peut assimiler au concept de « potentiel chimique » qui correspond à la concentration active d’une espèce chimique dans une phase donnée; celui-ci se définit comme le rapport entre la concentration (ou la pression) d’une substance (Ci) et une concentration standard (Co). En fait, la fugacité est une mesure du potentiel chimique sous la forme d’une pression ajustée et se définit par une équation simple :
où la fugacité (f) est égale à la concentration (Ci) du composé divisée par une constante (Z). L’entité mathématique Z est appelée « la constante de fugacité » et dépend de la nature de la molécule et de ses propriétés physiques intrinsèques, de la température, de la pression et du compartiment (phase) étudié. Ainsi, chaque composé aura tendance à s’accumuler dans les compartiments ayant une valeur de Z élevée et une faible fugacité relative. Comme il s’agit d’un modèle à l’équilibre, il faut que la fugacité soit égale dans tous les compartiments au moment d’atteindre l’équilibre. C’est donc la concentration de la molécule qui variera en fonction des paramètres environnementaux. On comprend qu’il devient alors possible de calculer la concentration du composé (Ci) dans chacune des phases en calculant la constante Z.
Un développement mathématique simple permet de démontrer que Z est directement dépendant du « coefficient de partage » entre deux phases. Par exemple, le calcul de Z pour l’eau est donné par :
où Kwa est le coefficient de partage entre l’eau et l’air et Za est la constante de fugacité dans l’air. Le développement de cette équation, en utilisant la thermodynamique des gaz, permet de démontrer que Kwa est simplement égale à l’inverse de la constante de la loi de Henry (Kwa = 1/H), une valeur qui mesure la solubilité d’un gaz dans un solvant liquide et est facilement accessible pour la grande majorité des composés organiques d’un intérêt environnemental.
Par ce développement, on voit aussi qu’il est facile d’ajouter d’autres compartiments comme le sédiment ou même un organisme biologique. Dans ces cas, le coefficient de partage octanol-eau (log Kow) et d’autres coefficients de partage (les facteurs de bioconcentration, les coefficients d’adsorption) seront utilisés suivant la même approche. La dernière étape à franchir est la fixation des frontières du modèle. Il faut fixer le nombre et la taille des compartiments, les conditions de pression et de température dans ces mêmes compartiments et enfin déterminer la quantité totale de la substance devant être introduite dans le modèle, c’est-à-dire la source de la contamination.
Une application récente du modèle a été présentée par une équipe pluridisciplinaire (BLAIS et al., 2007) et avait pour objectif de déterminer l’importance relative du transport de certains contaminants par les espèces migratrices comme les saumons et les oiseaux : un processus appelé « biovecteur » et particulièrement important pour les régions polaires où on trouve généralement peu ou pas de sources directes de contamination. À partir d’un modèle comptant quatre (4) compartiments abiotiques (sol, sédiment, eau et air) et pas moins de huit (8) compartiments biotiques (plancton, benthos, petits et grands poissons, saumons, oiseaux, ours et macrofaune terrestre), les auteurs ont calculé la fugacité d’un composé type (similaire à un BPC) avec une masse moléculaire de 300 g/mol, un Kwa de 0,01, un log Kow de 6 et une dégradabilité négligeable. En se basant sur des données de terrain, ils ont ensuite introduit le contaminant type dans les compartiments air, eau et saumon en respectant des concentrations réalistes pour une région nordique. Les résultats montrent que la migration des saumons contribue annuellement, et de façon cyclique, à augmenter la présence du contaminant dans tous les compartiments, sauf l’eau et l’air, à cause des très grands volumes considérés. L’apport annuel est faible mais constant et contribue donc à augmenter le bruit de fond de ce contaminant dans un environnement où les autres sources sont négligeables. Les données de terrain avaient déjà confirmé un biotransport des BPC par les saumons qui remontent les rivières pour frayer (KRÜMMEL et al., 2003) et on pourrait croire que le modèle vient simplement confirmer des informations déjà acquises. En fait, le modèle sert à synthétiser toutes les informations empiriques disponibles pour contraindre le modèle et permet surtout d’identifier les variables clés comme les variations saisonnières et les changements dans le contenu lipidique des divers compartiments biotiques (BLAIS et al., 2007).
Le modèle de la fugacité et les autres modèles de dispersion et de transport des contaminants organiques dans les divers compartiments de l’environnement contribuent de façon tangible aux études de risques puisque le risque environnemental est lié à la concentration d’un ou plusieurs contaminants dans un compartiment donné et à leur biodisponibilité respective. Tous les modèles sont cependant tributaires de la qualité des données qui sont utilisées pour les calculs et sont généralement construits pour répondre à des processus globaux à grande échelle comme dans l’exemple ci-haut. Plus l’échelle sera réduite en matière de volume des compartiments, plus il sera nécessaire de disposer de données qui seront spécifiques à la région étudiée, sinon le modèle ne pourra fournir que des généralités peu utiles à la gestion environnementale. Autre limitation importante, les modèles thermodynamiques doivent considérer le système à l’équilibre et ne peuvent pas incorporer les cinétiques lentes d’un processus évolutif, qu’il soit chimique ou biologique.
4.3 Biodisponibilité et bioaccessibilité
Afin d’illustrer certaines des difficultés rencontrées avec les modèles de dispersion, nous prendrons pour exemple le cas des HAP pour lequel un grand nombre d’études sont parues au cours des cinq dernières années. Les problèmes soulevés par la présence des HAP dans l’environnement sont multiples à cause, d’une part, de la multitude de sources anthropiques et naturelles de cette vaste famille de molécules toxiques et, d’autre part, de la variation dans le temps de leur biodisponibilité. Il faut aussi prendre en compte la biodégradabilité de la plupart de ces molécules par les organismes vivants et l’éventuelle persistance et la toxicité de leurs métabolites.
Nos travaux les plus récents ont porté sur la séquestration (ou l’emprisonnement) des HAP par divers substrats naturels ou anthropiques que l’on désigne souvent sous le nom générique de « géosorbants » (particules minérales ou organiques capables d’adsorber des molécules hydrophobes). Les mécanismes d’adsorption et de séquestration ont commencé à être décrits au milieu des années 1990 lorsqu’un modèle conceptuel et quelques mécanismes de base (LUTHY et al., 1997) ont été proposés pour expliquer les nombreuses observations de laboratoire et de terrain, apparaissant souvent contradictoires les unes par rapport aux autres. Le modèle proposait une première adsorption rapide du composé hydrophobe sur diverses surfaces organiques (matière organique amorphe, résidus de combustion, phases liquides non aqueuses) et minérales (argiles, phases minérales diverses), suivie d’une deuxième phase de séquestration à l’intérieur même des particules de géosorbants. Par contre, le modèle ne fournissait aucune indication sur la vitesse des divers mécanismes d’adsorption et de séquestration.
En utilisant des sédiments déjà contaminés par des HAP industriels et des traceurs deutérés, nous avons démontré la possibilité de décrire la cinétique d’adsorption et de séquestration de quelques HAP par un modèle simple à trois compartiments avec une cinétique de premier ordre en deux étapes non réversibles (Figure 5). En posant comme hypothèse de travail qu’en tout temps t, la quantité totale du contaminant introduit dans le modèle [W]0 demeure constante (pas de biodégradation, ni d’évaporation), on peut écrire :
où [W], [Ads] et [Seq] représentent les concentrations dans chacun des compartiments. On peut ensuite résoudre les trois équations de vitesse du modèle et parvenir à une expression mathématique pour chacun des compartiments en fonction des constantes de vitesse k1 et k2 (BRION et PELLETIER, 2005). En examinant les données expérimentales obtenues pour le compartiment Ads pour sept HAP et trois sédiments différents, nous avons constaté que la forme générale de la régression déterminée au mieux avec nos données correspondait très bien à l’équation théorique de ce compartiment :
où y, a, b et c sont respectivement associés aux paramètres [Ads], [W]0, les constantes k1 et k2. La détermination des constantes de vitesse a permis ensuite de calculer [Seq] et d’estimer qu’il faut généralement entre trois et cinq jours pour qu’un HAP dissous au départ atteigne sa concentration maximale dans le compartiment Ads (facilement extractible avec une solution tensioactive) et qu’il fallait de 17 à 21 jours pour que le HAP quitte ce compartiment et devienne peu disponible dans le compartiment Seq. Le modèle nous indique aussi des différences très significatives entre les HAP en fonction de leur taille et de leur structure. Par contre, on trouve relativement peu de différences entre les sédiments quant aux vitesses de séquestration. Malgré les nombreuses contraintes imposées par un modèle aussi simple, ces résultats apparaissent importants à plusieurs points de vue :
le modèle cinétique est applicable à tout contaminant chimique, incluant les métaux;
le modèle peut être développé en y introduisant la notion de réversibilité des réactions (voir BRION, 2003);
le modèle confirme la présence d’au moins deux compartiments dans le processus de séquestration;
les données montrent que l’adsorption de molécules hydrophobes sur les particules sédimentaires naturelles n’est pas instantanée mais requiert plusieurs jours pour atteindre sa concentration maximale dans le compartiment facilement accessible;
la séquestration s’étale sur plusieurs semaines et n’atteint jamais 100 % de la quantité de HAP introduit dans le système;
les HAP relâchés dans l’environnement aquatique restent donc facilement biodisponibles pendant des semaines avant d’être véritablement séquestrés.
Figure 5
Schéma du modèle cinétique à trois compartiments pour étudier la cinétique de séquestration des HAP par les particules sédimentaires en suspension. Les symboles sont expliqués dans le texte.
A three-compartment kinetic model representing the kinetics of PAH (polyaromatic hydrocarbons) sequestration by sedimentary particles in suspension. The symbols are described in the text.

Étant donné que la séquestration est un processus relativement lent et qu’il dépend fortement de la composition même du géosorbant et des propriétés du contaminant hydrophobe, il devient très difficile, voire impossible, d’établir par une approche mathématique les coefficients de partage eau-sédiment et sédiment-biote qui sont essentiels au modèle de la fugacité décrit précédemment. Il reste donc une approche expérimentale qui implique l’utilisation d’organismes vivants (bactéries, vers, bivalves, poissons, etc.) pour effectuer des tests de bioaccumulation et de métabolisation des HAP et en déduire leur biodisponibilité. On peut aussi tenter de simplifier le problème par une approche chimique qui permettrait de mimer l’action d’un organisme vivant dans le sédiment, comme un polychète. La biodisponibilité se définit comme la fraction du contaminant disponible pour l’ingestion, l’accumulation ou l’assimilation par un organisme. Mais cette définition crée une certaine confusion et il peut devenir nécessaire d’établir une différence entre « biodisponibilité » et « bioaccessibilité » (SEMPLE et al., 2004). Ainsi, ce qui est biodisponible (au sens strict d’une molécule en solution capable de traverser une membrane biologique) n’est pas nécessairement bioaccessible par l’organisme qui ne pourrait pas y accéder physiquement à cause, par exemple, de micropores trop étroits. Cette distinction apparemment subtile prend toute son importance quand vient le temps de comparer les méthodes chimiques dites « d’extraction sélective » avec les données biologiques de bioaccumulation. Que mesure-t-on au juste avec ces méthodes chimiques?
Nous avons commencé le travail en développant une nouvelle méthode d’extraction sélective utilisant un tensioactif de haut poids moléculaire (Brij®700, 4680 g mol‑1) et en comparant les résultats d’extraction des HAP avec trois autres méthodes d’extraction bien établies et aussi avec la bioaccumulation après 28 jours par le polychète Nereis virens pour les sédiments marins et l’oligochète Lumbriculus variegatus pour les sédiments lacustres (BARTHE et PELLETIER, 2007). Dans un premier temps, l’expérience montre que le tensioactif Brij®700 parvient assez bien à prédire le niveau total des HAP dans les deux vers pour les sédiments fortement contaminés (25 à 5 700 µg g‑1 sec) alors que le butanol et l’hydroxypropyl‑β‑cyclodextrine (HPDD) seraient les meilleurs prédicteurs pour les HAP tirés des sédiments faiblement contaminés (0,06 à 1,1 µg g‑1 sec). Des difficultés apparaissent pour chacune des méthodes chimiques quand on examine en détail leur performance pour chacun des 12 HAP analysés. On observe une grande dispersion des données et un net effet de la concentration des HAP dans le sédiment sur la capacité prédictive de la méthode chimique. En fait, toutes les extractions chimiques sous-estiment la bioaccumulation de la plupart des HAP par un facteur de 5 à 10 fois pour les sédiments faiblement contaminés. Ce travail fait ressortir, pour la première fois avec une preuve expérimentale solide, que la concentration des HAP dans le sédiment influence leur biodisponibilité relative pour les vers. Contrairement à nos attentes, la bioaccessibilité (biologique) est plus élevée que la biodisponibilité (chimique) pour les sédiments faiblement contaminés et n’ayant pas reçu d’apport industriel provenant des alumineries. Nous avons consolidé ces résultats avec deux autres études faisant intervenir des échantillonneurs passifs (BARTHE et al., 2008; BREEDVELD et al., 2007). Il s’agit non plus d’une extraction solide/liquide mais d’un échange solide/liquide/solide où l’on met en contact pendant plusieurs jours un sédiment en suspension dans l’eau avec un échantillonneur passif comme un morceau de polyoxyméthylène (POM) ou des billes de Tenax® que l’on peut ensuite extraire et en déduire la quantité de « HAP libres » et donc biodisponibles (JONKER et KOELMANS, 2001). Cette approche permet de calculer les facteurs d’accumulation sédiment-biote (BSAF) et de les comparer avec ceux obtenus avec un ver ou une autre technique chimique. De nouveau, les résultats montrent une grande disparité entre les échantillonneurs et entre les sédiments testés. Toutes les méthodes chimiques surestiment la biodisponibilité des HAP ayant un log Kow > 6 mais sous-estiment grandement la bioaccessibilité des HAP se trouvant dans les sédiments marins et lacustres faiblement contaminés (BARTHE et al., 2008). Pour les HAP de poids moléculaire élevé et fortement hydrophobes, il est possible que leur bioaccessibilité soit réduite par leur encombrement stérique, d’une part, et leur tendance à se dissoudre dans les phases liquides non aqueuses comme le goudron et les acides humiques condensés, d’autre part. En ce qui concerne l’hyper-accessibilité des HAP dans les sédiments peu contaminés, l’hypothèse la plus plausible est que les HAP s’y trouvent principalement associés à des particules organiques digestibles (microorganismes et débris biologiques) que les vers ingèrent et métabolisent. Les HAP sont alors transférés dans le ver et lentement métabolisés. D’autres travaux en cours sur les métabolites 9‑hydroxyphenanthrene et 1‑hydroxypyrène, nous montrent que les vers sont en mesure de produire et de bioaccumuler ces métabolites tout au long de la période d’exposition (BARTHE, résultats non publiés).
L’exemple des HAP montre toute la complexité et la variabilité des mécanismes chimiques et biologiques qui ont cours lors de la prise en change et de la métabolisation d’un contaminant organique par un invertébré. La même remarque s’applique aux autres grandes familles de contaminants hydrophobes comme les BPC, DDT et PBDE. Dès lors, on comprend les limitations importantes que rencontrent les modélisateurs dans l’application de modèles de dispersion des contaminants à une échelle régionale. De ce fait, on comprend avec quelle prudence il faut aborder les conclusions des évaluations de risques qui dépendent de ces modèles.
4.4 Le cas particulier des organométaux
Comme nous l’avons précédemment mentionné, un organométal doit comporter au moins un lien covalent entre un métal et un atome de carbone (parfois avec le soufre ou l’azote) et, de ce fait, il partage les propriétés d’un métal et celles d’une molécule organique. Il y a peu d’organométaux qui survivent longuement au formidable pouvoir oxydant de l’environnement, mais ceux qui y résistent possèdent alors de puissantes propriétés toxiques, ce qui en font des substances à haut risque pour l’environnement aquatique.
Le plus simple des organométaux est le méthylmercure qui n’est pas d’origine anthropique mais dont la synthèse par les bactéries est stimulée par la présence du mercure inorganique dans l’eau et les sédiments (GILMOUR et HENRY, 1991). Le mercure est apporté encore aujourd’hui par la combustion du charbon et les feux des forêts, les opérations minières (LACERDA, 1997) et le lessivage d’anciens sites industriels contaminés au mercure au cours des années 1950 à 1980 (LODENUS, 1998). Les données fournies par Environnement Canada (COSSA et al., 1997) montrent que le fleuve Saint-Laurent à la hauteur de la ville de Québec transportait encore en 1996 plus de 1 000 kg de mercure dissous et particulaire par année. Le méthylmercure est liposoluble et fortement neurotoxique (CHEUK et WONG, 2006; DEBES et al., 2006)). Les sources principales du méthylmercure pour les humains sont le poisson et les fruits de mer (GRANDJEAN et al., 1992) et l’écotoxicologie aquatique du méthylmercure demeure d’une grande actualité même si des accidents de l’ampleur de Minamata (EKINO et al., 2007) risquent peu de se reproduire.
Nos travaux au Saguenay (Québec) au début des années 1990 ont été les premiers à démontrer la présence significative de méthylmercure à l’interface oxique/anoxique d’un sédiment fortement contaminé au mercure en milieu marin (GAGNON et al., 1996). Depuis cette époque, plusieurs équipes ont travaillé à établir la distribution du méthylmercure entre les divers compartiments du milieu aquatique et pour certains sites particuliers (MASON et SULLIVAN, 1997; MAURY-BRACHET et al., 2006; PARK et al., 2007). La formation et le devenir du méthylmercure dans le biote sont très complexes et dépendent d’un grand nombre de facteurs comme la lumière incidente, le pH, la composition du carbone organique dissous, la diète et le taux de croissance des espèces impliquées dans le réseau trophique (WATRAS et al., 1998). Il n’existe pas encore de modèles prédictifs de la biodisponibilité et de la biomagnification du méthylmercure (CHEN et al., 2005) qui soient applicables à divers milieux aquatiques comme précédemment décrits pour les molécules organiques persistantes.
Le deuxième cas important est celui du tétraéthylplomb, un organométal de fabrication industrielle qui a été longtemps ajouté à l’essence pour en améliorer les performances. En conditions environnementales, le tetraéthylplomb (une molécule neutre et hydrophobe) se dégrade lentement pour former deux composés ioniques, le triéthylplomb et le diéthylplomb. Même si les essences au plomb ont été progressivement retirées du marché depuis les années 1970, on a trouvé des alkylplombs dans tous les compartiments de l’environnement à partir de l’air ambiant (HEWITT et HARRISON, 1987), en passant par les sédiments et les bivalves (CHAKRABORTI et al., 1989; MIKAC et al., 1996) et jusqu’au cerveau humain (NIELSEN et al., 1978)! Lors d’une analyse détaillée des contaminants, nous avons trouvé des quantités appréciables de tétraéthylplomb dans les sédiments accumulés à proximité du quai de Baie‑Sainte-Catherine à l’embouchure du fjord du Saguenay (Québec), indice clair de la persistance de ce composé dans les sédiments de zones portuaires et de marinas (PELLETIER, résultats non publiés). À moyen terme, le tétraéthylplomb ne devrait plus représenter une menace environnementale (SEYFERT, 2003), mais l’essence au plomb est encore utilisée dans certains pays en développement. Peu d’efforts ont été consentis pour la modélisation du comportement des alkylplombs dans les divers compartiments environnementaux.
Le troisième groupe d’organométaux méritant une attention toute particulière des écotoxicologues est celui des composés organostanniques. L’étain est un bon donneur d’électrons et forme facilement des liens covalents avec des atomes de carbone. La liaison Sn-C résiste très bien à l’hydrolyse et assure la persistance de ces composés dans les sols et sédiments, et en particulier dans les zones anoxiques et froides des régions nordiques (VIGLINO et al., 2004).
Le tributylétain (TBT) et le triphénylétain ont été synthétisés par milliers de tonnes depuis leur apparition sur le marché au milieu des années 1970 (BENNETT, 1996). Ils sont surtout utilisés comme biocides dans les peintures anti-salissures qui recouvrent les coques des navires marchands et des forces navales de plusieurs pays. Les effets toxiques dévastateurs des butylétains sur les bivalves (ALZIEU, 2000) et les gastéropodes (TEN HALLERS-TJABBES et al., 2003) ont été rapidement reconnus dès le début des années 1980 mais il a fallu plus de 20 ans avant qu’une action internationale ne se dessine et qu’un moratoire sur l’utilisation des peintures à l’étain ne soit proposé pour l’ensemble de la flotte mondiale (IMO, 2001). L’application de ce moratoire reste fragile et sujet à la bonne volonté de plusieurs pays, dont le Canada et les États-Unis. Le tributylétain demeure la seule substance hautement toxique (FENT, 1996) introduite volontairement et massivement dans l’environnement aquatique (essentiellement le milieu marin) pour solutionner un problème industriel causé par des organismes biologiques.
Les butylétains étant surtout présents dans les sédiments des ports commerciaux et des marinas, nous avons d’abord été surpris de trouver de fortes quantités de tri‑ et di‑butylétains dans les sédiments et le biote du fjord du Saguenay au début des années 2000 (VIGLINO et al., 2004 et 2005; VIGLINO et PELLETIER, 2006). Les butylétains ont été retrouvés en abondance (jusqu’ à 288 ng g‑1sec dans les sédiments de surface) en amont du fjord dans la baie des Ha! Ha!, avec une décroissance graduelle vers l’aval, montrant bien que la source principale se situait dans le secteur de Port-Alfred où des navires marchands viennent régulièrement décharger des minerais et embarquer des produits industriels. L’analyse de plusieurs carottes de sédiment a permis de calculer pour le TBT un coefficient de partition eau-sédiment élevé (log Kws = 4) et un très faible flux de diffusion eau-sédiment (1,54 x 10‑3 ng cm‑2 an‑1), ce qui explique largement sa préservation dans le sédiment ainsi que sa biodisponibilité. Comparativement, nous avions estimé le flux vertical du mercure inorganique à 20 ng cm‑2 an‑1 et celui du méthylmercure à 0,07 ng cm‑2 an‑1, des valeurs nettement supérieures à celle du TBT. Nous avons estimé que le temps de demi-vie du TBT dans la couche anoxique du sédiment du fjord était d’environ 87 ans alors qu’il n’était que d’environ huit ans dans la couche oxique de surface.
La contamination des espèces marines (seston, poissons, crustacés, échinodermes, bivalves et gastéropodes) était ubiquiste dans tout l’écosystème du fjord avec des facteurs d’accumulation biote-sédiments (« biota-sediment accumulation factor » ou BSAF) très variables de 0,32 à 11 (calculés pour les butylétains totaux), mais sans bioamplification importante entre les niveaux trophiques (VIGLINO et al., 2005). La mesure de l’imposex (ELLIS et PATTISSINA, 1990) chez Buccinum undatum a montré qu’une forte proportion des gastéropodes males de la baie des Ha! Ha! était affectée par la malformation de leurs organes génitaux. L’examen détaillé des proportions relatives de TBT et de ses deux métabolites le DBT (dibutylétain) et le MBT (monobutylétain) a permis de mettre en évidence des différences significatives entre les espèces et entre les organes d’une même espèce. Les poissons montraient une forte proportion de TBT, malgré leur capacité reconnue de dégrader les butylétains via le cytochrome P450, alors que le gastéropode B. undatum, clairement affecté par la toxicité du TBT, montrait pourtant une proportion souvent inférieure à 40 % de TBT dans ses tissus.
La somme de nos travaux dans le Saguenay et d’autres travaux menés dans le Saint-Laurent (MICHAUD et PELLETIER, 2006) et ailleurs dans l’est du Canada (ST-JEAN et al., 1999) nous ont amenés aux quelques conclusions suivantes :
la géomorphologie du fjord du Saguenay en fait un environnement semi-fermé très propice à la rétention des contaminants, dont les butylétains;
la principale route de prise en charge des butylétains par les invertébrés est la nourriture alors que les poissons montrent une certaine contribution par l’eau;
les capacités de dégradation et d’élimination des butylétains mesurées en terrain semblent en contradiction avec les données de laboratoire, en particulier pour les invertébrés fouisseurs comme les vers et les gastéropodes;
tout comme pour les HAP, les BSAF sont plus élevés pour des sédiments faiblement contaminés (estuaire du Saint-Laurent) comparativement à ceux fortement contaminés (fjord du Saguenay);
aucun des modèles décrits précédemment n’est en mesure de prédire le comportement des butylétains en milieu nordique.
5. Un nouveau défi : les nano-particules industrielles
Dans les paragraphes précédents, nous avons souvent fait allusion aux phases dissoute et particulaire pour décrire l’état physique d’un contaminant. Cette dualité « dissous/particulaire » est au coeur des modèles de biodisponibilité aussi bien pour les métaux que pour les substances organiques. Il n’existe pas de frontière théorique entre le dissous et le particulaire et les chimistes utilisent généralement une définition empirique pour distinguer dans quelle phase se trouve un matériau donné. Par exemple, on admet que tout ce qui passe à travers un filtre de 0,20 ou encore de 0,45 µm est dissous ou de « nature colloïdale ». Cette frontière pragmatique cause cependant de sérieux maux de tête quand on aborde le nouveau domaine de la nanotechnologie et des nanoparticules industrielles (voir BUFFLE, 2006 ainsi que LEAD et WILKINSON, 2006 pour quelques définitions utiles). En fait, les nanoparticules naturelles (suies, aérosols, virus, zéolites, argiles, oxydes métalliques, etc.) sont connues depuis longtemps mais la nouveauté ici est que la nanotechnologie permet de fabriquer en quantité industrielle des nanoparticules de dimension, de forme et de composition contrôlées pour des applications industrielles multiples (cosmétique, catalyse hétérogène, électronique, optique, et applications biomédicales, parmi tant d’autres). Le fait qu’il soit maintenant possible de fonctionnaliser la surface d’une particule de quelques nanomètres et que cette nanoparticule puisse diffuser ou être transportée à travers une membrane biologique à la manière d’un métal dissous, si elle est chargée, ou d’une molécule organique, si elle est neutre, pose toute la question du risque pour la santé humaine et l’environnement de ces nanomatériaux (LEROUEIL et al., 2007; MOORE, 2006). Une toute nouvelle branche de la toxicologie est en train de se développer : la « nanotoxicologie », dont le champ d’activité porte essentiellement sur l’étude du comportement des nanoparticules et de leurs effets sur les cellules et les tissus biologiques (DONALDSON et al., 2004; OBERDÖRSTER et al., 2005). Par extension au domaine des sciences environnementales, on pourra parler de nanotoxicologie environnementale et même peut-être d’éco-nanotoxicologie pour désigner le secteur de recherche portant sur les effets toxiques et les risques environnementaux des nanoparticules pour l’environnement aquatique et les écosystèmes. À peine quelques laboratoires de par le monde ont commencé à explorer la toxicité des nanoparticules métalliques comme le cuivre (GRIFFITT et al., 2007), l’argent (LEE et al., 2007) et l’oxyde de titane (FEDERICI et al., 2007) ainsi que les nanotubes de carbone (TEMPLETON et al., 2006).
Les défis que posent la nanotoxicologie environnementale sont multiples et pour la plupart inexplorés. Le tout premier est d’ordre analytique puisqu’il n’existe pratiquement aucune méthode pour quantifier la présence de nanoparticules dans un tissu biologique, un sédiment ou même dans des eaux naturelles (NOVACK et BUCHELI, 2007). Même si on parvient à bien voir les nanoparticules en microscopie électronique et en microscopie optique pour les particules marquées par un fluorophore, il est très difficile de séparer les nanoparticules de la matrice environnementale (CORNELISSEN et al., 2005) et donc de les caractériser une fois qu’elles ont atteint le milieu aquatique. Le deuxième défi est celui du comportement environnemental des nanoparticules qui peuvent atteindre l’environnement sous diverses formes : libres (particules individuelles); agrégées (plusieurs nanoparticules formant des microparticules); dispersées au sein d’une matrice organique comme un polymère; ou encore fonctionnalisées (particules individuelles ou agrégées).
Les nanoparticules ont généralement tendance à former des agrégats en milieu aqueux à cause de leur faible solubilité (les nanoparticules organiques) ou de leur charge de surface (les nanoparticules minérales ou métalliques). Cependant, on découvre peu à peu qu’il existe un équilibre entre les agrégats et les nanoparticules libres parce que celles-ci peuvent se recouvrir de composés naturels qui favorisent la dispersion (HYUNG et al., 2007). Il faut voir ici qu’une nanoparticule peut devenir un vecteur pour le transport membranaire d’une molécule toxique (NOWACK et BUCHELI, 2007). Le troisième défi est aussi d’ordre analytique mais concerne les tests de toxicité. Comment déterminer les effets toxiques de nanoparticules sur un organisme aquatique ou terrestre si on contrôle mal l’état physico-chimique des particules (agrégat ↔ nanoparticules libres ↔ nanoparticules fonctionnalisées ou enveloppées)? Les risques de faux positifs ou de faux négatifs semblent importants dans ce cas. Nous croyons que des modèles cellulaires devront être développés spécifiquement pour les nanoparticules.
Le quatrième défi qui nous apparaît comme majeur est celui de la modélisation du devenir des nanoparticules en utilisant une approche comme la fugacité. La dualité dissous/particulaire pour la matière dans un compartiment environnemental donné cause un problème de la même importance que celui de la dualité onde/particule pour la lumière. Par exemple, comment caractériser une matière dite colloïdale qui devient « nanoparticulaire » sous l’effet d’un changement du pH ou de la force ionique de la solution? Il ne s’agit plus du transport d’un compartiment vers un autre (caractérisé par une constante de fugacité ou une constante de vitesse) mais d’une modification de phase à l’intérieur du même compartiment. Non seulement une nanoparticule peut changer de phase, mais elle peut aussi changer de taille (agrégat) et changer de nature en s’appropriant des ions métalliques et/ou des molécules organiques en solution. Dans les faits, une nanoparticule métallique chargée et obéissant au modèle BLM peut devenir une nanoparticule organique et neutre se comportant comme une molécule organique traversant passivement les membranes et pouvant être métabolisée.
6. Conclusion
On voit par l’exemple des nanoparticules que l’avenir est à l’intégration des concepts et modèles utilisés en chimie environnementale pour les métaux et les molécules organiques afin d’arriver à une approche « universelle » du comportement et du devenir des xénobiotiques (toute substance étrangère et potentiellement dommageable au bon fonctionnement cellulaire) dans l’environnement aquatique et les écosystèmes. L’organisme SETAC (Society of Environmental Toxicology and Chemistry) a initié une réflexion en ce sens lors d’un atelier tenu en mai 2003 où plusieurs experts ont opté pour une approche globale qui permette d’ordonner les risques provenant à la fois des métaux et des molécules organiques pour un milieu aquatique donné (PETERS et al., 2005). Étant donné que l’approche classique PBT (persistance, bioaccumulation et toxicité) se révèle peu efficace pour les métaux (débordement) et parfois trompeuse pour les molécules organiques (séquestration) et les organométaux (biodégradation), les auteurs ont convenu de développer un concept unifié appelé UWM (Unit World Model) qui est une extension du modèle de la fugacité. L’UWM représente un environnement bien défini pouvant recevoir une charge critique d’un contaminant qui sera ensuite distribué dans divers compartiments (eau, biote, poissons, oiseaux) en tenant compte de tous les facteurs physiques, chimiques et biologiques pouvant influencer ce transfert et sa rétention. Comme nous l’avons vu précédemment, cette approche est déjà utilisée pour les substances organiques (BLAIS et al., 2007) mais demeure un défi important pour les métaux (HARVEY et al., 2007). Le principal intérêt du modèle est de dépasser les considérations particulières sur la bioaccumulation ou la toxicité et de fournir une expression globale du risque sous la forme d’une « charge critique » dans un compartiment biotique bien défini. Il suffit ensuite d’utiliser un indicateur de toxicité (LC50 ou un niveau d’effet sous-létal) pour évaluer le risque encouru. Les risques combinés provenant d’une exposition simultanée d’un compartiment à un ou plusieurs métaux et un ou plusieurs composés organiques pourraient, en principe, être évalués par ce type de modèle à la condition de disposer des indicateurs de toxicité appropriés. Cependant, quel que soit le niveau de sophistication qu’atteindront les modèles en voie de développement, leurs performances seront toujours dépendantes de la qualité des données qui y seront introduites.
En conclusion de cette synthèse, il nous apparait que l’avenir de l’écotoxicologie aquatique est tributaire des quatre éléments fondamentaux suivants :
l’obtention de données de terrain et de laboratoire de qualité toujours croissante par l’application de nouvelles techniques analytiques et de monitoring;
une compréhension approfondie des mécanismes de toxicité au niveau moléculaire et cellulaire;
le développement de modèles théoriques et empiriques toujours plus performants;
le développement d’indicateurs écosystémiques capables de fournir une image globale de la qualité d’un environnement aquatique, quelle que soit sa complexité.
Parties annexes
Remerciements
Les auteurs tiennent à remercier sincèrement les étudiants, les stagiaires postdoctoraux et le personnel de recherche pour leur intérêt profond pour l’écotoxicologie et pour leur contribution inestimable à l’avancement des connaissances dans un domaine au coeur des préoccupations du monde moderne. Les auteurs remercient également le programme des Chaires de recherche du Canada, la Fondation canadienne pour l’innovation (FCI), le Conseil de recherches en sciences naturelles et en génie du Canada (CRSNG) et le Fonds québécois de recherche sur la nature et les technologies (FQRNT) pour leur support financier apporté au cours des dernières années, un support qui a permis de développer d’importantes infrastructures de recherche et de fournir un soutien financier à tout le personnel étudiant, technique et professionnel qui compose nos équipes respectives.
Références bibliographiques
- ALZIEU, C. (2000). Impact of tributyltin on marine invertebrates. Ecotoxicol. 9, 71-76.
- ANKLEY, G.T., G.P. DASTON, S.J. DEGITZ, N.D. DENSLOW, R.A. HOKE, S.W. KENNEDY, A.L. MIRACLE, E.J. PERKINS, J. SNAPE, D.E. TILLITT, C.R. TYLER et D. VERSTEEG (2006). Toxicogenomics in regulatory ecotoxicology. Environ. Sci. Technol., 40, 4055-4065.
- BARTHE, M. et E. PELLETIER (2007). Comparing bulk extraction methods for chemically available polycyclic hydrocarbons with bioaccumulation in worms. Environ. Chem., 4, 271-283.
- BARTHE, M., E. PELLETIER, G.D. BREEDVELD et G. CORNELISSEN (2008). Passive samplers versus surfactant extraction for the evaluation of PAH availability in sediments with variable levels of contamination. Chemosphere (sous presse).
- BENNETT, R.F. (1996). Industrial manufacture and applications of tributyltin compounds. Dans : Tributyltin: case study of an environmental contaminant. de Mora, S. (Éditeur). Cambridge Environmental Chemistry Series, Cambridge University Press, Cambridge, Grande-Bretagne, Chap. 2, pp. 21-61.
- BLAIS, J.M., R.W. MACDONALD, D. MACKAY, E. WEBSTER, C. HARVEY et J.P. SMOL (2007). Biologically mediated transport of contaminants to aquatic systems. Environ. Sci. Technol., 41, 1075-1084.
- BONDARENKO, S., J. GAN, D.L. HAVER et J.N. KABASHIMA (2004). Persistence of selected organophosphate and carbamate insecticides in waters from a coastal watershed. Environ. Toxicol. Chem., 23, 2649-2654.
- BOUDOU, A. et F. RIBEYRE (1989). Aquatic ecotoxicology: fundamental concepts and methodologies. CRC Press, Boca Raton, FL, États-Unis, 332 p.
- BREEDVELD, G.D., E. PELLETIER, R. ST-LOUIS et G. CORNELISSEN (2007). Sorption characteristics of polycyclic hydrocarbons in aluminum smelter residues. Environ. Sci. Technol., 41, 2542-2547.
- BRION, D. (2003). La cinétique de séquestration des HAP dans les sédiments marins et lacustres. Mémoire de maîtrise, Université du Québec à Rimouski, Québec, 94 p.
- BRION, D. et E. PELLETIER (2005). Modelling PAHs adsorption and sequestration in freshwater and marine sediments. Chemosphere, 61, 867-876.
- BUFFLE, J. (2006). The key role of environmental colloids/nanoparticles for the sustainability of life. Environ. Chem., 3, 155-158.
- BUTLER, G.C. (1978). Principles of Ecotoxicology. John Wiley & Sons, Chichester, Grande-Bretagne, 350 p.
- CAHILL, T.M., I. COUSINS et D. MACKAY (2003). General fugacity-based model to predict the environmental fate of multiple chemical species. Environ. Toxicol. Chem., 22, 483-493.
- CAMPBELL, P.G.C. (1995). Interactions between trace metals and organisms: a critique of the free-ion activity model. Dans : Metal Speciation and Bioavailability in Aquatic Systems. TESSIER A. et D. TURNER (Éditeurs). J. Wiley & Sons, Chichester, Grande-Bretagne, pp. 45-102.
- CAMPBELL, P.G.C., P.M. CHAPMAN et B.A. HALE (2006). Risk assessment of metals in the environment. Issues Environ. Sci. Technol., 21, 102-131.
- CAMPBELL, P.G.C. et Y. COUILLARD (2004). Prise en charge et détoxication des métaux chez les organismes aquatiques. Dans : Écotoxicologie moléculaire - Principes fondamentaux et perspectives de développement. PELLETIER E., P.G.C. CAMPBELL, et F. DENIZEAU (Éditeurs). Les Presses de l’Université du Québec, Ste-Foy, Québec, Canada, pp. 9‑61.
- CAMPBELL, P.G.C., A. GIGUÈRE, E. BONNERIS et L. HARE (2005). Cadmium-handling strategies in two chronically exposed indigenous freshwater organisms - the yellow perch (Perca flavescens) and the floater mollusc (Pyganodon grandis). Aquat. Toxicol., 72, 83-97.
- CAMPBELL, P.G.C., A. HONTELA, J.B. RASMUSSEN, A. GIGUÈRE, A. GRAVEL, L. KRAEMER, J. KOVECSES, A. LACROIX, H. LEVESQUE et G.D. SHERWOOD (2003). Differentiating between direct (physiological) and food-chain mediated (bioenergetic) effects on fish in metal-impacted lakes. Human Ecol. Risk Assess., 9, 847-866.
- CAMPBELL, P.G.C., L.D. KRAEMER, A. GIGUÈRE, L. HARE et A. HONTELA (2008). Subcellular distribution of cadmium and nickel in chronically exposed wild fish: inferences regarding metal detoxification strategies and implications for setting water quality guidelines for dissolved metals. Human Ecol. Risk Assess., 14, 290-316.
- CARSON, R. (1962). Silent Spring. Houghton Mifflin, New York, NY, États-Unis, 378 p.
- CHAKRABORTI, D., W. DIRKX, R. VAN CLEUVENBERGEN et F. ADAMS (1989). Speciation of ionic alkyllead in potable water and soil. Sci. Tot. Environ., 84, 249-257.
- CHEN, C.Y., R.S. STEMBERGER, N.C. KANNAN, B.M. MAYES et C.L. FOLT (2005). Patterns of Hg bioaccumulation and transfer in aquatic food webs across multi-lake studies in the northeast US. Ecotoxicol., 14, 135-147.
- CHEUK, D.K. et V. WONG (2006). Attention-deficit hyperactivity disorder and blood mercury level: A case-control study in Chinese children. Neuropediatrics, 37, 234-240.
- CORNELISSEN, G., ¸Ö GUSTAFSSON, T.D. BUCHELI, M.T.O. JONKER, A.A. JOELMANS et P.C.M. VAN NOORT (2005). Extensive sorption of organic compounds to black carbon, coal, and kerogen in sediments and soils: mechanisms and consequences for distribution, bioaccumulation, and bioaccumulation. Environ. Sci. Technol., 39, 6881-6895.
- COSSA, D., T.T. PHAM, B. RONDEAU, B. QUÉMERAIS, S. PROULX et C. SURETTE (1997). Bilan massique des contaminants chimiques dans le fleuve Saint-Laurent. Environnement Canada – Région du Québec, Conservation de l’environnement. Centre Saint-Laurent. Rapport scientifique et technique, 117 p.
- DEBES, F., E. BUDTZ-JORGENSEN, P. WEIHE, R.F. WHITE et P. GRANDJEAN (2006). Impact of prenatal methylmercury exposure on neurobehavioral function at age 14 years. Neurotoxicol. Teratol., 28, 363-375.
- DONALDSON, K., V. STONE, C.L. TRAN, W. KREYLING et P.L.A. BORM (2004). Nanotoxicology. Occup. Environ. Med., 61, 727-728.
- DUDAL, Y. et F. GÉRARD (2004). Accounting for natural organic matter in aqueous chemical equilibrium models: a review of the theories and applications. Earth-Sci. Rev., 66, 199-216.
- EKINO, S., M. SUSA, T. NINOMIYA, K. IMAMURA et T. KITAMURA (2007). Minamata disease revisited: an update on the acute and chronic manifestations of methyl mercury poisoning. J. Neurol. Sci., 262, 131-144.
- ELLIS, D.V. et L.A. PATTISSINA (1990). Widespread neogastropod imposex: a biological indicator of global TBT contamination. Mar. Pollut. Bull., 21, 248-253.
- FEDERICI, G., B.J. SHAW et R.D. HANDY (2007). Toxicity of titanium dioxide nanoparticles to rainbow trout (Oncorhynchus mykiss): gill, injury, oxidative stress, and other physiological effects. Aquat. Toxicol., 84, 415-430.
- FENT, K. (1996). Ecotoxicology of organotin compounds. Crit. Rev. Toxicol., 26, 1-117.
- FISHER, N.S. et S.E. HOOK (2002). Toxicology tests with aquatic animals need to consider the trophic transfer of metals. Toxicol., 181, 531-536.
- GAGNON, C., A. MUCCI et E. PELLETIER (1997). Behaviour of anthropogenic mercury in coastal marine sediments. Mar. Chem., 59,159-176.
- GAGNON, C., E. PELLETIER et A. MUCCI (1996). Diagenetic behaviour of methylmercury in organic-rich coastal sediments. Limnol. Oceanogr., 41, 428-434.
- GARRETT, R.G. (2000). Natural sources of metals to the environment. Human Ecol. Risk Assess., 6, 945-963.
- GIGUÈRE, A., P.G.C. CAMPBELL, L. HARE et P. COUTURE (2006). Sub-cellular partitioning of cadmium, copper, nickel and zinc in indigenous yellow perch (Perca flavescens) sampled along a polymetallic gradient. Aquat. Toxicol., 77, 178-189.
- GILMOUR, C. et E. HENRY (1991). Mercury methylation in the aquatic systems affected by acid deposition. Environ. Pollut., 71, 131-169.
- GONZALEZ, P., M. BAUDRIMONT, A. BOUDOU et J.P. BOURDINEAUD (2006). Comparative effects of direct cadmium contamination on gene expression in gills, liver, skeletal muscle and brain of the zebra fish (Danio rerio). Biometals, 19, 225-235.
- GRANDJEAN, P., P. WEIHE, P.J. JORGENSEN, T. CLARKSON, E. CERNICHIARI et T. VIDERO (1992). Impact of maternal seafood diet on fetal exposure to mercury, selenium, and lead. Arch. Environ. Health, 47, 185-195.
- GRAVEL, A., P.G.C. CAMPBELL et A. HONTELA (2005). Disruption of the hypothalamo-pituitary-interrenal axis in 1+ yellow perch (Perca flavescens) chronically exposed to metals in the environment. Can. J. Fish. Aquat. Sci., 62, 982-990.
- GRIFFITT, R., R. WEIL, K.A. HYNDMAN, N.D. DENSLOW, K. POWERS, D. TAYLOR et D.S. BARBER (2007). Exposure to copper nanoparticles causes gill injury and acute lethality in zebrafish (Danio rerio). Environ. Sci. Technol., 41, 8178-8186.
- HALE, R.C., M. ALAEE, J.B. MANCHESTER-NEESVIG, H.M. STAPLETON et M.G. IKONOMOU (2003). Polybrominated diphenyl ether flame retardants in the North American environment. Environ. Internat., 29, 771‑779.
- HANSEN, L.J., L.H. SCHWACKE, G.B. MITCHUM, A.A. HOHN, R.S. WELLS et E.S. ZOLMAN (2004). Geographic variation in polychlorinated biphenyl and organochlorine pesticide concentrations in the bubbler of bottlenose dolphins from the US Atlantic coast. Sci. Tot. Environ., 319, 147-172.
- HARVEY C., D. MACKAY et E. WEBSTER (2007). Can the Unit World Model concept be applied to hazard assessment of both organic chemicals and metal ions? Environ. Toxicol. Chem., 26, 2129-2142.
- HEWITT, C.N. et R.M. HARRISON (1987). Atmospheric concentrations and chemistry of alkyllead and environmental alkylation of lead. Environ. Sci. Technol., 21, 260-272.
- HOGSTRAND, C., S. BALESARIA et C.N. GLOVER (2002). Application of genomics and proteomics for study of the integrated response to zinc exposure in a non-model fish species, the rainbow trout. Comp. Biochem. Physiol. Part B, 133, 523-535.
- HOOK, S.E. et N.S. FISHER (2001). Sublethal effects of silver in zooplankton: Importance of exposure pathways and implications for toxicity testing. Environ. Toxicol. Chem., 20, 568-574.
- HYUNG, H., J.D. FORTNER, J.B. HUDGHES et J.H. KIM (2007). Natural organic matter stabilizes carbon nanotubes in the aqueous phase. Environ. Sci. Technol., 41, 179-184.
- IMO, 2001. International Convention on Control of Harmful Anti-fouling Systems on Ships. International Maritime Organization. http://www.imo.org/ (consultation le 30 décembre 2007).
- JONKER, M.T.O. et A.A. KOELMANS (2001). Polyethylene solid-phase extraction as a partitioning method for hydrophobic organic chemicals in sediment and soot. Environ. Sci. Technol., 35, 3742-3749.
- KAMUNDE, C., C. CLAYTON et C.M. WOOD (2002). Waterborne vs. dietary copper uptake in rainbow trout and the effects of previous waterborne copper exposure. Amer. J. Physiol. - Regul. Integr. Comp. Physiol. 283, R69-R78.
- KLINCK, J.S., W.W. GREEN, R.S. MIRZA, S.R. NADELLA, M. CHOWDHURY, C.M. WOOD et G.G. PYLE (2007). Branchial cadmium and copper binding and intestinal cadmium uptake in wild yellow perch (Perca flavescens) from clean and metal-contaminated lakes. Aquat. Toxicol., 84, 198-207.
- KRAEMER, L.D., P.G.C. CAMPBELL et L. HARE (2008). Modeling Cd accumulation in indigenous yellow perch (Perca flavescens). Can. J. Fish.Aquat. Sci., sous presse.
- KRÜMMEL, E., I. GREGORY-EAVES, R.W. MACDONALD, L.E. KIMPE, M. DEMERS, J.P.SMOL, B. FINLEY et J.M. BLAIS (2003). Delivery of pollutants by spawning salmon. Nature, 425, 255-256.
- LACERDA, L. (1997). Global mercury emissions from gold and silver mining. Wat. Air Soil Pollut., 97, 209-221.
- LEAD, J.R. et K.J. WILKINSON (2006). Aquatic colloids and nanoparticles: current knowledge and future trends. Environ. Chem., 3, 159-171.
- LEE, K.J., P.D. NALLATHAMBY, L.M. BROWNING, C.J. OSGOOD et X.-H. N. XU (2007). In vivo imaging of transport and biocompatibility of single silver nanoparticules in early development of zebrafish embryos. ACSNano, 1, 133‑143.
- LEROUEIL, P.R., S. HONG, A. MECKE, J.R. BAKER et M.M. BANASAK HOLL (2007). Nanoparticle interaction with biological membranes: does nanotechnology present a Janus face? Acc. Chem. Res., 40, 325-342.
- LEVESQUE, H., J. DORVAL, G.J. VAN DER KRAAK, P.G.C. CAMPBELL et A. HONTELA (2003). Hormonal, morphological and physiological responses of yellow perch (Perca flavescens) to chronic environmental metal exposures. J. Toxicol. Environ. Health, 66, 657-676.
- LODENUS, M. (1998). Dry and wet deposition of mercury near a chlor-alkali plant. Sci. Tot. Environ., 213, 53-56.
- LUOMA, S.N. et P.S. RAINBOW (2005). Why is metal bioaccumulation so variable? Biodynamics as a unifying principle. Environ. Sci. Technol., 39, 1921-1931.
- LUTHY, R.G., G.R. AIKEN, M.L. BRUSSEAU, S.D. CUNNINGHAM, P.M. GSCHWEND, J.J, PIGNATELLO, M. REINHARD, S.J. TRAINE, W.J. WEBER et J.C. WESTALL (1997). Sequestration of hydrophobic organic contaminants by geosorbents. Environ. Sci. Technol., 31, 3341‑3347.
- MACKAY, D. (1979). Finding fugacity feasible. Environ. Sci. Technol., 13, 1218-1223.
- MACKAY, D. (1991). Multimedia Environmental Models. The Fugacity Approach. Lewis Publishers, Chelsea, Michigan, États-Unis, 257 p.
- MACKAY, D. et A. FRASER (2000). Bioaccumulation of persistent organic chemicals: mechanisms and models. Environ. Pollut., 110, 375-391.
- MACKAY, D., L.S. MCCARTY et M. MACLEOD (2001). On the validity of classifying chemicals for persistence, bioaccumulation, toxicity, and potential for long-range transport. Environ. Toxicol. Chem., 20, 1491-1498.
- MACKAY, D. et S. PATERSON (1982). Fugacity revisited. Environ. Sci. Technol., 16, 654-660.
- MASON, A.Z. et K.D. JENKINS (1995). Metal detoxification in aquatic organisms. Dans : Metal Speciation and Bioavailability in Aquatic Systems. TESSIER A. et D. TURNER (Éditeurs). John Wiley & Sons, Chichester, Grande-Bretagne, pp. 479-608.
- MASON, A.Z., S.D. STORMS et K.D. JENKINS (1990). Metalloprotein separation and analysis by directly coupled size exclusion high-performance liquid chromatography inductively coupled plasma mass spectroscopy. Anal. Biochem., 186, 187‑201.
- MASON, R. et K. SULLIVAN (1997). Mercury in Lake Michigan. Environ. Sci. Technol., 31, 942-947.
- MAURY-BRACHET, R., G. DURRIEU, Y. DOMINIQUE et A. BOUDOU (2006). Mercury distribution in fish organs and food regimes: significant relationships from twelve species collected in French Guiana (Amazonian basin). Sci. Tot. Environ., 368, 262-270.
- MERSIE, W., C. MCNAMEE, C. SEYBOLD, J. WU et D. TIERNEY (2004). Degradation of metolachlor in bare and vegetated soils and in simulated water-sediment systems. Environ. Toxicol. Chem., 23, 2627-2632.
- MEYER, J.S., W.J. ADAMS, K.V. BRIX, S.N. LUOMA, D.R. MOUNT, W.A. STUBBLEFIELD et C.M. WOOD (2005). Toxicity of Dietborne Metals to Aquatic Biota. Society of Environmental Toxicology and Chemistry, SETAC Press, Pensacola, FL, États-Unis, 303 p.
- MICHAUD, H. et E. PELLETIER (2006). Sources and fate of butyltins in the St. Lawrence Estuary ecosystem. Chemosphere, 64, 1074-1082.
- MIKAC, N., M. BRANICA, Y. WANG et R.M. HARRISON (1996). Organolead compounds in the mussels (Mytilus galloprovincialis) from the Eastern Adriatic Coast. Environ. Sci. Technol., 30, 499-508.
- MILNE, C.J., D.G. KINNIBURGH et E. TIPPING (2001). Generic NICA-Donnan model parameters for proton binding by humic substances. Environ. Sci. Technol., 35, 2049‑2059.
- MILNE, C.J., D.G. KINNIBURGH, W.H. VAN RIEMSDIJK et E. TIPPING (2003). Generic NICA-Donnan model parameters for metal-ion binding by humic substances. Environ. Sci. Technol., 37, 958-971.
- MOORE, M.N. (2006). Do nanoparticles present ecotoxicological risks for the health of aquatic environments? Environ. Internat., 32, 967-976.
- NIELSEN, T., K.A. JENSEN et P. GRANDJEAN (1978). Organic lead in normal human brains. Nature, 274, 602-603.
- NIYOGI, S. et C.M. WOOD (2004). Biotic Ligand Model, a flexible tool for developing site-specific water quality guidelines for metals. Environ. Sci. Technol., 38, 6177-6192.
- NOVACK, B. et T.D. BUCHELI (2007). Occurrence, behavior and effects of nanoparticles in the environment. Environ. Pollut., 150, 5-22.
- NRIAGU, J.O. (1989). A global assessment of natural sources of atmospheric trace metals. Nature, 338, 47-49.
- OBERDÖRSTER, G., E. OBERDÖSTER et J. OBERDÖSTER (2005). Nanotoxicology: an emerging discipline evolving from studies of ultrafine particles. Environ. Health Persp., 113, 823-839.
- OLENDRZYNSKI, K., S. ANDERBERG, J. BARTNICKI, J.M. PACYNA et W. STIGLIANI (1996). Atmospheric emissions and depositions of cadmium, lead, and zinc in Europe during the period 1955-1987. Environ. Rev., 4, 300-320.
- PACYNA, E.G., J.M. PACYNA, F. STEENHUISEN et S. WILSON (2006). Global anthropogenic mercury emission inventory for 2000. Atmos. Environ., 40, 4048-4063.
- PACYNA, J.M., M.T. SCHOLTZ et Y.-F.A. LI (1995). Global budget of trace metal sources. Environ. Rev., 3, 145‑159.
- PAQUIN, P.R., J.W. GORSUCH, S. APTE, G.E. BATLEY, K.C. BOWLES, P.G.C. CAMPBELL, C.G. DELOS, D.M. DI TORO, R.L. DWYER, F. GALVEZ, R.W. GENSEMER, G.G. GOSS, C. HOGSTRAND, C.R. JANSSEN, J.C. MCGEER, R.B. NADDY, R.C. PLAYLE, R.C. SANTORE, U. SCHNEIDER, W.A. STUBBLEFIELD, C.M. WOOD et K.B. WU (2002). The biotic ligand model: a historical overview. Comp. Biochem. Physiol. C - Toxicol. Pharmacol., 133, 3-35.
- PAQUIN, P.R., R.C. SANTORE, K.J. FARLEY, C. KAVVADAS, K.B. WU, K. MOONEY et D.M. DI TORO (2003). A Review: Exposure, Bioaccumulation, and Toxicity Models for Metals in Aquatic Systems. SETAC Press, Pensacola, FL, États-Unis, 160 p.
- PARK, M.S., J.R. BURGESS, D.C.. EVERS et C.S. LOFTIN (2007). Mercury contamination of biota from Acadia National Park, Maine: A review. Environ. Monit. Assess., 126,105-115.
- PELLETIER, E., P.G.C. CAMPBELL et F. DENIZEAU (2004). Écotoxicologie moléculaire - Principes fondamentaux et perspectives de développement. Les Presses de l’Université du Québec, Quebec, Canada, 450 p.
- PETERS A., W.J. ADAMS, M.L. DIAMOND, W. DAVISON, D.M. DI TORO, P.J. DOYLE, D. MACKAY, J. NRIAGU, C. PTACEK, J.M. SKEAFF, E. TIPPING et H. WAETERSCHOOT (2005). Integrated approach for hazard assessment of metals and inorganic metal substances: the Unit World Model approach. Dans : Assessing the hazard of metals and inorganic metal substances in aquatic and terrestrial systems. ADAMS W.J. et P.M. CHAPMAN (Éditeurs), CRC Press, Taylor and Francis Group, Boca Raton, FL, États-Unis, pp. 11‑54.
- PIERRON, F., M. BAUDRIMONT, P. GONZALEZ, J.P. BOURDINEAUD, P. ELIE et J.C. MASSABUAU (2007). Common pattern of gene expression in response to hypoxia or cadmium in the gills of the European glass eel (Anguilla anguilla). Environ. Sci. Technol., 41, 3005-3011.
- RAMADE, F. (1979). Écotoxicologie. Masson S.A., Paris, France, 227 p.
- RASMUSSEN, J.B., J.M. GUNN, A. ILES, A. GAGNON, A. LACROIX, P.G.C. CAMPBELL et A. HONTELA (2008). Direct and indirect (foodweb-mediated) effects of metal exposure on the growth of yellow perch (Perca flavescens): Implications for ecological risk assessment. Human Ecol.Risk Assess., sous presse.
- REIMANN, C. et R.G. GARRETT (2005). Geochemical background - concept and reality. Sci. Tot. Environ., 350, 12‑27.
- RICHARDSON, G.M., I.A. MITCHELL, M. MAH-PAULSON, T. HACKBARTH et R.G. GARRETT (2003). Natural emissions of mercury to the atmosphere in Canada. Environ. Res., 11, 17-36.
- ROSS, P.S., G.M. ELLIS, G.M. IKONOMOU, L.G. BARRETT-LENNARD et R.F. ADDISON (2000). High PCB concentrations in free-ranging Pacific killer whales, Orcinus orca: effects of age, sex and dietary preference. Mar. Pollut. Biol., 40, 504-515.
- SCHWARZENBACH, R.P., P.M. GSCHWEND et D.M. IMBODEN (1993). Environmental Organic Chemistry. John Wiley and Sons Ltd., New York, NY, États-Unis, 550 p.
- SEMPLE, K.T., K.J. DOICK, K.C. JONES, P. BURAUEL, A. CRAVEN et H. HARMS (2004). Defining bioavailability and bioaccessibility of contaminated soil and sediment is complicated. Environ. Sci. Technol., 38, 228A-231A.
- SEYFERT, D. (2003). The rise and fall of tetraethyllead. Organometallics, 22, 5154-5178.
- SHEEHAN, P.J., D.R. MILLER, G.C. BUTLER et P. BOUDREAU (1984). The effects of pollutants at the ecosystem level. John Wiley & Sons, Chichester, Grande-Bretagne, 443 p.
- SIGG, L., F. BLACK, J. BUFFLE, J. CAO, R. CLEVEN, W. DAVISON, J. GALCERAN, P. GUNKEL, E. KALIS, D. KISTLER, M. MARTIN, S. NOEL, Y. NUR, N. ODZAK, J. PUY, W. VAN RIEMSDIJK, E. TEMMINGHOFF, M.L. TERCIER-WAEBER, S. TOEPPERWIEN, R.M. TOWN, E. UNSWORTH, K.W. WARNKEN, L.P. WENG, H.B. XUE et H. ZHANG (2006). Comparison of analytical techniques for dynamic trace metal speciation in natural freshwaters. Environ. Sci. Technol., 40, 1934-1941.
- STEINBERG, C.E.W. et M. ADE (2005). Ecotoxicology, Where do you come from and where do you go? Environ. Sci. Pollut. Res., 12, 245-246.
- ST-JEAN, S.D., S.C. COURTENAY, E. PELLETIER et R. ST-LOUIS (1999). Tributyltin concentrations in sediments and blue mussels (Mytilus edulis) of the southern Gulf of St. Lawrence, Canada. Environ. Technol., 20, 181-189.
- TEMPLETON, R.C., P.L. FERGUSON, K.M. WASHBURN, W.A. SCRIVENS et G.T. CHANDLER (2006). Life-cycle effects of single-walled carbon nanotubes (SWNTs) on an estuarine meiobenthic copepod. Environ. Sci. Technol., 40, 7387-7393.
- TEN HALLERS-TJABBES, C.C., J.W. WEGENER, B. VAN HATTUM, J.F. KEMP, E. TEN HALLERS, T.J. REITSEMA et J.P. BOON (2003). Imposex and organotin concentrations in Buccinum undatum and Neptunea antique from the North Sea: relationship to shipping density and hydrographical conditions. Mar. Environ. Res., 55, 203-233.
- TIPPING, E. (1998). Humic ion-binding Model VI: an improved description of the interactions of protons and metal ions with humic substances. Aquat. Geochem., 4, 3-48.
- TIPPING, E. (2002). Cation binding by humic substances. Cambridge University Press, Cambridge, Grande-Bretagne, 444 p.
- TURNER, D.R. (1995). Problems in trace metal speciation modeling. Dans : Metal Speciation and Bioavailability in Aquatic Systems. TESSIER A. et D. TURNER (Éditeurs). J. Wiley & Sons, Chichester, Grande-Bretagne, pp. 149-203.
- UNSWORTH, E.R., K.W. WARNKEN, H. ZHANG, W. DAVISON, F. BLACK, J. BUFFLE, J. CAO, R. CLEVEN, J. GALCERAN, P. GUNKEL, E. KALIS, D. KISTLER, H.P. VAN LEEUWEN, M. MARTIN, S. NOEL, Y. NUR, O. ODZAK, J. PUY, W. VAN RIEMSDIJK, L. SIGG, E. TEMMINGHOFF, M.-L. TERCIER-WAEBER, S. TOEPPERWEIN, R.M. TOWN, L. WENG et H. XUE (2006). Model predictions of metal speciation in freshwaters compared to measurements by in situ techniques. Environ. Sci. Technol., 40, 1942-1949.
- US EPA (1998). Guidelines for Ecological Risk Assessment. US Environmental Protection Agency, Risk Assessment Forum No. EPA/630/R095/002F, Washington, DC, États-Unis, 188 p.
- US EPA (2007). Framework for Metals Risk Assessment. U.S. Environmental Protection Agency, Office of the Science Advisor, Risk Assessment Forum No. EPA 120/R-07/001, Washington, DC, États-Unis, 171 p.
- VELTMAN, K., J. HENDRIKS, M. HUIJBREGTS, P. LEONARDS, M. VAN DEN HEUVEL-GREVE et D. VETHAAK (2005). Accumulation of organochlorines and brominated flame retardants in estuarine and marine food chains: field measurements and model calculations. Mar. Pollut. Bull., 50, 2085-1102.
- VIGLINO, L., E. PELLETIER et R. ST-LOUIS (2004).Highly persistent butyltins in Northern marine sediments: a long-term threat for the Saguenay Fjord (Canada). Environ. Toxicol. Chem., 23, 2673-1681.
- VIGLINO, L., E. PELLETIER et L.E.J. LEE (2005). Butyltin species in benthic and pelagic organisms of the Saguenay Fjord (Canada) and imposex occurrence in the Common Whelk. Arch. Environ. Contam. Toxicol., 49, 1-17.
- VIGLINO, L. et E. PELLETIER (2006). Butylétains dans les eaux du fjord du Saguenay (Canada) : menace pour l’écosystème d’un milieu-semi-fermé. Rev. Sci. Eau, 19,11‑22.
- VIJVER, M.G., C.A.M. VAN GESTEL, R.P. LANNO, N.M. VAN STRAALEN et W.J.G.M. PEIJNENBURG (2004). Internal metal sequestration and its ecotoxicological relevance: a review. Environ. Sci. Technol., 38, 4705-4712.
- WALLACE, W.G., B.-G. LEE et S.N. LUOMA (2003). Subcellular compartmentalization of Cd and Zn in two bivalves. I. Significance of metal-sensitive fractions (MSF) and biologically detoxified metal (BDM). Mar. Ecol. Progr. Ser., 249, 183-197.
- WANG, W.-X. et P.S. RAINBOW (2006). Subcellular partitioning and the prediction of cadmium toxicity to aquatic organisms. Environ. Chem., 3, 395-399.
- WANIA, F. et D. MACKAY (1993). Global fractionation and cold condensation of low volatility organochlorine compounds in polar compounds. Ambio, 22, 10-18.
- WATRAS, C.J., R.C. BACK, S. HALVORSEN, R.J.M. HUDSON, K.A. MORRISON et S.P. WENTE (1998). Bioaccumulation of mercury in pelagic freshwater food webs. Sci. Tot. Environ., 219, 183-208.
- WRIGHT, D.A. et P.M. WELBOURN (2002). Environmental Toxicology. Cambridge University Press, Cambridge, Grande-Bretagne, 630 p.
Liste des figures
Figure 1
Prise en charge de métaux par des organismes aquatiques – mécanismes généraux : (i) diffusion passive de complexes métalliques lipophiles; (ii) transport facilité de cations; (iii) transport accidentel de cations liés à des anions.
Metal uptake by aquatic organisms – general mechanisms: (i) passive diffusion of lipophilic metal complexes; (ii) facilitated cation transport; (iii) accidental transport of cations bound to anions.
Figure 2
Modélisation biodynamique : représentation simplifiée (modèle à un compartiment) de l’accumulation et de l’élimination d’un contaminant par un organisme aquatique (LUOMA et RAINBOW 2005). Les paramètres du modèle sont expliqués dans le texte.
Biodynamic modeling: simplified representation (one compartment model) of the accumulation and elimination of a contaminant by an aquatic organism (LUOMA and RAINBOW, 2005). The model parameters are explained in the text.
Figure 3
Concentrations en Cd hépatique dans diverses fractions subcellulaires chez la perchaude jaune, Perca flavescens: (a) protéines thermosensibles, (b) organelles et (c) protéines thermostables. Les résultats sont présentés en fonction des concentrations hépatique totales en Cd dans les poissons récoltés dans des lacs le long d’un gradient de concentrations métalliques (CAMPBELL et al., 2008). Les données proviennent d’études spatiales en 2001 (cercles) et en 2003 (triangles). Dans le graphique (b), les cercles solides = lysosomes + microsomes; les cercles ouvertes = mitochondries. Chaque point correspond à la valeur moyenne pour les poissons récoltés à une seule station d’échantillonnage dans un lac donné. Figure adaptée de CAMPBELL et al. (2008), avec la permission de la maison de publication Taylor et Francis.
Concentrations of hepatic Cd in various subcellular fractions in yellow perch, Perca flavescens: (a) heat-denaturable proteins, (b) organelles and (c) heat-stable proteins. The results are presented as a function of total hepatic Cd in juvenile perch sampled along a metal concentration gradient (CAMPBELL et al., 2008). Data come from spatial studies in 2001 (circles) and 2003 (triangles). In panel (b), solid circles = lysosomes and microsomes, open circles = mitochondria. Each point represents data for individual fish collected at a single sampling station. Figure adapted from CAMPBELL et al. (2008), with the permission of the publisher, Taylor and Francis.
Figure 4
Structure générale des polybromo-diphényléthers (PBDE).
General structure of polybromo-diphenylethers (PBDE).
Figure 5
Schéma du modèle cinétique à trois compartiments pour étudier la cinétique de séquestration des HAP par les particules sédimentaires en suspension. Les symboles sont expliqués dans le texte.
A three-compartment kinetic model representing the kinetics of PAH (polyaromatic hydrocarbons) sequestration by sedimentary particles in suspension. The symbols are described in the text.
Liste des tableaux
Tableau 1
Comparaison – métaux versus contaminants organiques synthétiques (adapté de CAMPBELL et al., 2006).
Comparison of metals and synthetic organic contaminants.
Caractéristique |
Contaminants organiques de synthèse |
Métaux |
|---|---|---|
Origine |
anthropique, sauf pour les hydrocarbures aromatiques polycycliques; |
géogénique (naturellement présents); |
bruit de fond → 0 |
bruit de fond variable |
|
Devenir |
sujets à divers processus de dégradation (hydrolyse, photolyse, biodégradation); |
pas sujets à des processus de dégradation; infiniment persistant, |
demi-vies environnementales utiles; |
demi-vies environnementales sans signification; |
|
composé original → dérivé (changements irréversibles) |
changements de spéciation (réversibles) |
|
bioamplification commune |
bioamplification rare (exception = méthylHg) |
|
Interactions avec des cellules vivantes |
prise en charge se produisant normalement par diffusion à travers une barrière bilipidique; |
prise en charge se produit normalement par transport facilité; |
effets narcotiques communs et toxicité des métabolites |
toxicité spécifique au métal |
|
Fonction biologique |
aucune; mécanismes complexes de défense contre la bioaccumulation des composés toxiques |
souvent biologiquement essentiel; bioaccumulation un phénomène naturel |