Abstracts
Résumé
Le gène suppresseur de tumeurs p53 contrôle l’expression d’une collection de gènes réglant le cycle cellulaire et l’apoptose, empêchant ainsi la prolifération de cellules porteuses de dommages et de remaniements génétiques. Cependant, une série de travaux a mis en évidence un nouveau mécanisme, indépendant de l’activité de transactivateur de transcription, par lequel p53 participe au maintien de la stabilité du génome : la répression de la recombinaison homologue. Ce rôle nouvellement décrit prévient l’instabilité génétique due à un excès de recombinaison homologue associé aux stress génotoxiques. Il s’exerce directement par l’interaction de la protéine p53 avec les protéines de recombinaison homologue et avec les structures d’ADN intermédiaires. Le domaine central de p53 est impliqué dans l’interaction avec Rad51, à une étape précoce de la recombinaison homologue, et le domaine carboxyterminal de p53 est nécessaire à son interaction avec Rad54 et avec les intermédiaires de recombinaison homologue, à une étape tardive de la recombinaison homologue. L’implication potentielle de ce mécanisme parallèle de p53 pour la stabilité du génome, la spéciation et la protection tumorale sera discutée.
Summary
The tumor suppressor gene p53, which is the most frequently mutated gene in human tumors, controls cell cycle checkpoint and apoptosis via the transactivation of the transcription of a collection of genes. These activities avoid proliferation of cell bearing alteration of genetic material. However, like a two-edged sword, p53 can also directly participate to genome stability maintenance by repressing homologous recombination (HR), independently of the transactivation activity. This parallel activity allows to limit the deleterious consequences on an excess of HR. Beside genetic interactions, p53 protein physically interacts with both HR proteins and HR intermediates (heteroduplex and Holliday junctions). The core domain of p53 is required for interaction with Rad51 at an early step and the carboxy-terminal domain of p53 is involved in the interaction with Rad54 and HR intermediates, at a late step. We discuss here the putative consequences of this parallel activity of p53 on genome stability, speciation and tumor protection.
Article body
La transmission fidèle du patrimoine génétique exige la coordination d’un réseau de voies métaboliques comprenant le contrôle du cycle cellulaire, l’apoptose et la réparation de l’ADN. Cet équilibre permet d’éviter la prolifération de cellules dont l’ADN est endommagé, qui pourrait conduire à des transformations malignes. Le gène suppresseur de tumeurs p53 est le gène le plus fréquemment muté dans les cancers. En réponse à un stress génotoxique, la protéine p53 active l’expression d’une série de gènes de contrôle du cycle cellulaire et de l’apoptose [1]. Cette activité de transactivateur transcriptionel est généralement considérée comme l’activité canonique onco-suppressive de p53. Cependant, depuis quelques années, différents laboratoires ont mis en évidence un rôle additionnel de p53 dans le contrôle de la plasticité du génome, la régulation de la recombinaison homologue, indépendante de son activité de transactivation. La protéine p53 agit directement sur la recombinaison homologue grâce à des interactions physiques avec les protéines et les structures d’ADN intermédiaires de recombinaison homologue.
La recombinaison homologue, facteur de stabilité, de variabilité ou d’instabilité génétique
La recombinaison homologue est un processus fondamental conservé dans tous les organismes et impliqué dans le contrôle de la plasticité du génome ((→) m/s 1990, n° 1, p. 18). Le modèle le plus étayé est celui de la réparation d’une cassure double brin (CDB) (Figure 1).
Figure 1
Modèle de réparation des cassures double brin (CDB) par recombinaison homomogue.
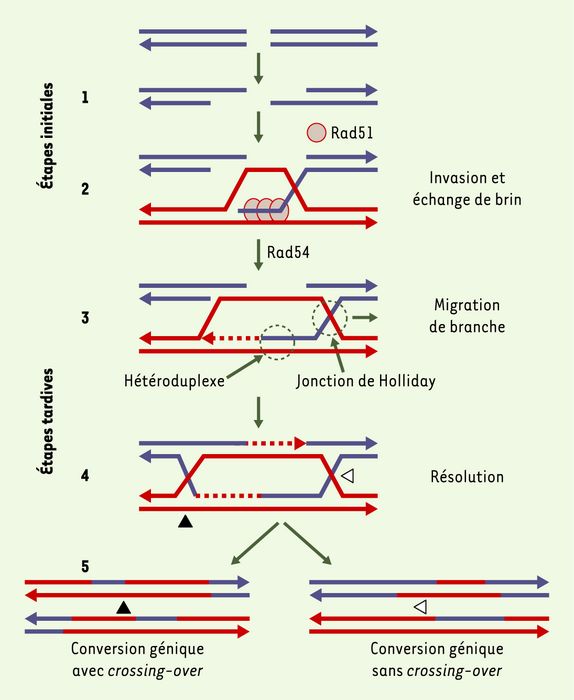
1. La CDB est métabolisée par une exonucléase qui produit des queues simple brin. 2. L’ADN simple brin envahit un ADN duplexe homologue intact. Cette étape requiert des séquences homologues, mais un polymorphisme limité est toléré par le processus : un échange de brin entre deux séquences non parfaitement homologues crée une molécule hybride porteuse de mésappariements de base, appelée hétéroduplexe, qui peut être prise en charge par le système de réparation des mésappariements de bases. Cette étape produit également des jonctions cruciformes (jonctions de Holliday) qui peuvent migrer (migration de branche), permettant ainsi l’élongation de la molécule hétéroduplexe. Cette étape initiale d’appariement, puis d’échange de brins, est catalysée par les protéines RecA chez la bactérie et Rad51 chez les eucaryotes. 3. Une polymérisation est déclenchée en utilisant l’extrémité 3’ du brin envahisseur comme amorce. Le brin d’ADN intact déplacé par la synthèse s’hybride alors avec la queue simple brin complémentaire, l’étape étant réalisée grâce à la protéine Rad54. Les cercles en pointillés indiquent les zones où la protéine p53 peut interagir directement avec l’intermédiaire de recombinaison homomogue. 4. Les brèches simple brin sont comblées par la polymérisation. 5. La conversion génique (transfert non réciproque d’information génétique ne modifiant pas la structure générale du chromosome) dépend de l’orientation de la réparation des mésappariements et de la séquence copiée dans la région de la CDB. Les jonctions de Holliday peuvent être résolues selon deux orientations alternatives (triangles noir ou blanc) : suivant l’orientation de résolution, les produits (conversion génique) seront associés on non à un crossing-over (échange réciproque d’information génétique) (d’après [38]).
La recombinaison homologue est considérée comme un système de réparation fidèle, car elle copie une séquence homologue intacte, ce qui lui permet d’assurer la stabilité du génome (réparation de l’ADN, redémarrage des fourches de réplication bloquées, ségrégation des chromosomes). Toutefois, l’échange entre séquences d’ADN homologues, mais non identiques, permet aussi à la recombinaison homologue de produire de la variabilité génétique (évolution moléculaire, diversification d’antigènes membranaires, genèse du répertoire immunitaire chez le poulet ou le lapin, brassage des allèles en méiose).
Un excès de recombinaison homologue peut cependant devenir délétère et conduire à des réarrangements chromosomiques, à des pertes d’hétérozygotie ou à des inactivations de gènes (Figure 2), toutes situations observées dans différentes maladies [2, 3]. Un contrôle précis de la recombinaison homologue est donc indispensable afin d’éviter une trop grande instabilité génétique. La protéine p53 participe directement à ce contrôle grâce à des interactions directes avec les protéines de recombinaison homologue (Rad51 et Rad54), ainsi qu’avec ses intermédiaires (hétéroduplexes et jonctions de Holliday).
Figure 2
Instabilité génétique associée à un excès de recombinaison homomogue.

A. Réarrangements chromosomiques résultant de crossing-over [39]. 1.Crossing-over inter-chromosomique ou entre chromatides soeurs décalées aboutissant à une amplification et à une délétion génique. 2.Crossing-over intrachromatidien entre séquence en répétition directe aboutissant à l’excision du fragment interne. 3.Crossing-over entre répétition en orientation inverse aboutissant à l’inversion du fragment interne. 4-5.Crossing-over interchromosomiques : selon l’orientation par rapport au centromère, les produits seront une translocation (4) ou un chromosome dicentrique et un chromosome acentrique (5). B. Conversion génique entre (1) deux hétéro-allèles aboutissant à une perte d’hétérozygotie ou entre (2) un pseudogène (flèche jaune) et un gène (flèche bleue), aboutissant à l’inactivation du gène. Les mutations transférées sont représentées par le trait rouge.
La protéine p53 inhibe la voie Rad51, indépendamment de son activité de transactivation
L’impact de p53 sur la recombinaison homologue a été étudié dans diverses lignées cellulaires. L’inactivation de p53 a été réalisée par des stratégies distinctes comprenant la délétion du gène p53, l’inactivation de la protéine p53 (mutation thermosensible, protéine antagoniste) ou l’expression d’un mutant dominant négatif. La recombinaison homologue a été mesurée dans ces modèles par l’analyse de la recombinaison homologue intermoléculaire entre deux génomes de virus SV40 défectueux [4], ou par l’étude de la recombinaison homologue intramoléculaire entre deux séquences répétées en tandem, intégrées dans le génome cellulaire [5-11]. Ces conclusions ont été récemment confirmées in vivo dans un modèle murin transgénique [4].
La protéine p53 affecte la voie de recombinaison homologue dépendante de Rad51
La recombinaison entre séquences en répétition directe peut se produire par plusieurs mécanismes, dont la conversion génique. En revanche, la recombinaison homologue entre séquences en répétition inverse ne se produit que par conversion génique. Or, l’inactivation de p53 stimule aussi bien la recombinaison homologue entre répétition directe qu’entre répétition inverse [11], ce qui montre que p53 affecte spécifiquement la conversion génique, un processus dépendant de Rad51 chez les mammifères [12]. Ces résultats identifient donc la voie Rad51 comme cible d’action de p53.
Les CDB de l’ADN stimulent fortement la recombinaison homologue chez les mammifères. Cependant, bien que les radiations ionisantes induisent des CDB, elles ne stimulent pas significativement la recombinaison homologue dans des cellules exprimant une p53 sauvage [11, 13]. En revanche, dans les cellules mutantes pour p53, la recombinaison homologue peut être fortement stimulée par les radiations ionisantes ou par une coupure enzymatique ciblée dans le substrat de recombinaison homologue [11, 14]. Comme la recombinaison homologue induite par les radiations ionisantes est dépendante de Rad51 [12], ces résultats désignent également la voie Rad51 comme cible de p53.
La recombinaison homologue permet de réactiver efficacement des fourches de réplication bloquées [15]. Des inhibiteurs de réplication comme l’hydroxyurée stimulent la recombinaison homologue dépendante de Rad51 [16], qui peut être réprimée par la protéine p53 sauvage [17, 18]. Récemment, il a été montré que la protéine BLM, inactivée dans le syndrome de Bloom, facilite le transport de la protéine p53 sur les fourches de réplication bloquées et son interaction avec la protéine Rad51 [19]. Un modèle a alors été suggéré, dans lequel la protéine p53 contrôle les processus alternatifs de maturation des fourches de réplication bloquées, favorisant les processus maintenant la stabilité du génome.
L’action de p53 sur la recombinaison homologue est indépendante de son activité de transactivation
L’étude de différentes formes de p53 (mutants ou variants d’épissage alternatif) ou de HDM2 (un inhibiteur de l’activité de transactivation de p53) montre que la répression de la recombinaison homologue par p53 est séparable de son rôle dans le contrôle du cycle cellulaire en G1, et plus généralement de son activité de transactivation [11, 20, 21]. Par ailleurs, la stimulation de la recombinaison homologue liée à une surexpression de Rad51 peut être annulée par la co-expression d’une p53 mutante, défectueuse pour la transactivation [22].
Cette fonction de p53 dans la répression de la recombinaison homologue dépendante de Rad51, et indépendante de son activité de transcription, est directe : in vitro, la protéine p53 inhibe la réaction d’échange de brin entre une molécule d’ADN simple brin et une molécule homologue double brin, catalysée par la protéine Rad51. De plus, une protéine p53 mutante qui n’inhibe pas la recombinaison homologue in vivo n’a pas d’effet sur les réactions étudiées in vitro [23].
Fondements moléculaires de la répression de la recombinaison homologue par p53
Le niveau d’expression de p53 est important pour la répression de la recombinaison homologue. Des cellules homozygotes ou hétérozygotes pour l’invalidation de p53 ont une haplo-insuffisance pour l’inhibition de la recombinaison homologue [10]. De plus, si une faible expression de p53 normale peut inhiber la recombinaison homologue [14], inversement, de faibles niveaux d’expression de p53 mutante à effet dominant négatif sont capables de stimuler la recombinaison homologue [11].
La protéine p53 possède un domaine central ainsi qu’un domaine d’oligomérisation, tous deux nécessaires à la répression de la recombinaison homologue [11, 20] (Figure 3A). En revanche, le domaine carboxyterminal n’est pas essentiel [20, 21], même s’il est impliqué dans la localisation de la protéine p53 sur les fourches de réplication bloquées [19], dans l’interaction avec la protéine de recombinaison Rad54 [22] et dans le métabolisme des intermédiaires de recombinaison [24, 25]. Le domaine central et le domaine carboxyterminal de p53 pourraient donc participer à la régulation de la recombinaison homologue par des voies métaboliques parallèles.
Figure 3
Structure de p53 et des protéines de recombinaison homologue Rad51 (humaine) et RecA (bactérienne).

A. Domaines fonctionnels de la protéine p53. SLN : signal de localisation nucléaire. B. Domaines fonctionnels de Rad51 et de RecA. Le domaine central, conservé au cours de l’évolution, est appelé « boucle RecA ». Les nombres indiquent la position des résidus d’acides aminés.
Interactions de p53 avec les protéines de recombinaison homologue Rad51 et Rad54
Les régions de p53 qui interagissent avec Rad51 sont localisées aux extrémités du domaine central [26] (Figure 3A). C’est aussi dans ce domaine que sont localisés les points chauds de mutation associés aux cancers. Le domaine d’interaction de p53 avec la protéine Rad54 est quant à lui localisé en son extrémité carboxyterminale [22].
Le domaine central de la protéine Rad51 (Figure 3B), qui contient des résidus fixant et hydrolysant l’ATP, est également impliqué dans l’homo-oligomérisation de la protéine, deux activités essentielles pour la recombinaison homologue. L’interaction de Rad51 avec la protéine p53 s’effectue par le biais d’une région comprise entre les deux « boîtes ATP » [26]. Il semble que l’interaction de p53 avec cette région affecte l’activité de Rad51 : ainsi, la répression de la recombinaison homologue par p53 n’est plus observée quand la protéine Rad51 utilisée est le mutant L186PRad51, incapable d’interagir avec p53 [22].
La protéine Rad51 contrôle le processus de recombinaison homologue en formant des complexes avec de nombreux partenaires, dont les rôles exacts restent à définir. En plus de ses cinq paralogues (Figure 4A), la protéine Rad51 interagit avec de nombreuses autres protéines également impliquées dans le contrôle de la recombinaison homologue, pour lesquelles des interactions physiques avec la protéine p53 ont été décrites (Figure 4B), ce qui suggère que la protéine p53 agit sur le processus de recombinaison homologue par l’intermédiaire d’un ou plusieurs partenaires de Rad51 [27].
Figure 4
Complexes protéiques de recombinaison homomogue chez les mammifères.

A. Les cinq paralogues (Xrcc2, Xrcc3, Rad51B, Rad51C et Rad51D) de la protéine Rad51 sont impliqués dans deux complexes différents. Aucune interaction physique entre ces paralogues et la protéine p53 n’a clairement été démontrée. B. La protéine Rad51 interagit physiquement avec les autres protéines contrôlant la recombinaison homomogue. Les protéines du complexe capables d’interagir physiquement avec p53 sont cerclées de rouge.
Interactions de p53 avec les intermédiaires de recombinaison homologue - complémentarité avec le système de réparation des mésappariements
Outre ses interactions avec les protéines de recombinaison homologue, p53 peut également fixer les molécules d’ADN intermédiaires de recombinaison homologue, les hétéroduplexes et les jonctions de Holliday. Ces caractéristiques fonctionnelles sont similaires à celles du système de réparation des mésappariements (MMR). En corrigeant les mésappariements présents dans les hétéroduplexes formés par l’échange de brins lors de la recombinaison homologue, le MMR inhibe la conversion génique.
Des expériences de recombinaison homologue in vivo menées sur différents types d’héréroduplexes ont montré une inhibition de la recombinaison homologue par la protéine p53, d’une façon dépendante du type de mésappariement ; cette inhibition est associée à l’affinité de la protéine p53 in vitro pour ces différents intermédiaires artificiels [28]. L’étape d’invasion de brin crée un intermédiaire à trois brins d’ADN (voir Figure 1) ; in vitro, p53 possède une bonne affinité pour ce type de structure, particulièrement s’il contient des mésappariements [28], et cet intermédiaire à trois brins serait un substrat privilégié de l’activité (controversée) exonucléase 3’->5’ de p53, peut-être stimulée par Rad51. Les hétéroduplexes seraient alors dégradés par p53 au cours de l’étape d’échange de brins catalysée par Rad51, aboutissant à une inhibition de la recombinaison homologue [29].
Si Msh2 (la protéine pivot du MMR) et p53 s’associent à l’ADN de manière stable dès lors qu’ils rencontrent un mésappariement [25], leurs affinités respectives pour les différents types de mésappariements sont opposées. En effet, les mésappariements les mieux reconnus par la protéine p53 sont les moins bien reconnus par la protéine Msh2, et inversement [30]. Les deux systèmes pourraient toutefois coopérer, car des interactions physiques entre p53 et MSH2 ont été observées [31], et Msh2 augmente la fixation de p53 sur l’ADN porteur de mésappariements [32].
À l’instar de Msh2, p53 peut fixer des jonctions de Holliday artificielles in vitro (voir Figure 1) [24], cette fixation étant stimulée par l’hétérodimère Msh2-Msh6 [32]. BLM et WRN (dont l’inactivation est responsable du syndrome de Werner) peuvent faire migrer les jonctions de Holliday. p53 interagit par son domaine carboxyterminal avec les hélicases BLM et WRN, responsables d’une migration des jonctions de Holliday ; in vitro, p53 inhibe l’activité de migration exercée par les hélicases sur une jonction synthétique cruciforme de 50 mers ((→) m/s 2002, n°1, p. 79).
Conclusions et spéculations
En tant que « gardienne du génome », p53 semble particulièrement « prudente », car elle multiplie systématiquement les systèmes de sauvegarde de la stabilité du génome : p53 agit ainsi indirectement, sur le contrôle du cycle cellulaire et l’apoptose via son activité de transactivation, mais aussi directement, en réprimant le mécanisme de recombinaison homologue ((→) m/s 2000, n°14, p. 1387). Ces deux rôles poursuivent le même but, la prévention de l’instabilité génétique, et pourraient donc représenter des systèmes de sauvegarde parallèles. Par ailleurs, p53 utilise deux stratégies complémentaires pour contrôler la recombinaison homologue, chacune impliquant un domaine différent : interaction avec Rad51, à une étape précoce, via son domaine central et, à une étape tardive, interaction avec Rad54 et les intermédiaires de recombinaison homologue via son domaine carboxyterminal.
Le système MMR et p53 présentent de nombreuses similitudes. Tous deux protègent l’organisme contre le développement de tumeurs et pourraient agir de façon complémentaire et coordonnée pour empêcher une instabilité génétique liée à un excès de recombinaison homologue [27]. Ces caractéristiques autorisent à s’interroger sur l’implication de l’activité anti-recombinaison homologue de p53 dans les différents processus fondamentaux discutés ci-dessous.
Recombinaison entre séquences répétées
La recombinaison entre des séquences répétées représente un danger potentiel pour l’intégrité du génome. Cependant, cette recombinaison est limitée par la divergence de séquence entre ces répétitions. En effet, un événement efficace de recombinaison homologue nécessite un segment minimal d’homologie (MEPS, minimal efficiency processing segment) ininterrompue d’une taille comprise entre 200 et 300 paires de base (pb) chez les mammifères [33]. Une étude systématique a montré que p53 crée un seuil de besoins en homologie pour la recombinaison homologue [9], p53 étant même encore plus efficace à réprimer la recombinaison homologue avec des séquences homologues inférieures à 200 pb [14]. D’après ces différents travaux, p53 pourrait participer au maintien de la stabilité du génome en contrôlant le MEPS [27].
Spéciation
La recombinaison entre des génomes de deux espèces différentes est limitée par la divergence des séquences, ce qui participe au maintien de la barrière génétique. Or, cette barrière génétique peut être abolie par l’inactivation du MMR, comme cela a pu être montré avec les bactéries Escherichia coli et Salmonella typhimurium, pourtant séparées de 100 à 150 millions d’années dans l’évolution et présentant 15 % à 20 % de divergence dans leurs génomes [34]. Si la protéine p53 empêchait la recombinaison homologue entre séquences divergentes, d’une manière complémentaire et parallèle au système MMR, il serait alors possible qu’elle participe aussi à la spéciation chez les organismes supérieurs.
Protection tumorale
L’activité de transactivation de la protéine p53 a un rôle essentiel dans deux processus de prévention tumorale, au moins : le contrôle du cycle cellulaire et l’apoptose. Un modèle transgénique murin défectueux pour l’activité de transactivation de p53 a d’ailleurs montré l’importance de cette activité pour la protection tumorale [35] ; cependant, ces animaux développent moins de tumeurs que des animaux ayant une délétion totale de p53, et présentent un spectre de tumeurs différent. Par ailleurs, l’inhibition chimique de l’activité de transactivation de p53 n’augmente pas la tumorigenèse [36]. Enfin, des souris dont le gène p21 a été invalidé et défectueuses pour l’arrêt en G1 ne développent pas plus de tumeurs que les souris sauvages [37]. Ces résultats suggèrent l’existence d’un mécanisme additionnel à la transactivation pour la protection tumorale par p53 : le contrôle d’un excès de recombinaison homologue, empêchant une trop grande instabilité génétique, pourrait être un bon candidat pour cette activité, même si la part respective de la transactivation et celle de l’inhibition de la recombinaison homologue sur le processus de protection tumorale doivent être évaluées plus directement. Cependant, la mise en évidence d’un rôle direct de la protéine p53 dans le contrôle de la plasticité du génome éclaire une dimension essentielle des fonctions de la protéine p53 dans la réponse cellulaire aux dommages de l’ADN.
Appendices
Références
- 1. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997 ; 88 : 323-31.
- 2. Amor M, Parker KL, Globerman H, et al. Mutation in the CYP21B gene (Ile-172-Asn) causes steroid 21-hydroxylase deficiency. Proc Natl Acad Sci USA 1988 ; 85 : 1600-4.
- 3. Purandare SM, Patel PI. Recombination hot spots and human disease. GenomeRes 1997 ; 7 : 773-86.
- 4. Wiesmuller L, Cammenga J, Deppert WW. In vivo assay of p53 function in homologous recombination between simian virus 40 chromosomes. J Virol 1996 ; 70 : 737-44.
- 5. Bishop AJ, Hollander MC, Kosaras B, et al. Atm-, p53-, and Gadd45a-deficient mice show an increased frequency of homologous recombination at different stages during development. Cancer Res 2003 ; 63 : 5335-43.
- 6. Meyn MS, Strasfeld L, Allen C. Testing the role of p53 in the expression of genetic instability and apoptosis in ataxia-telangiectasia. Int J Radiat Biol 1994 ; 66 : S141-9.
- 7. Bertrand P, Rouillard D, Boulet A, et al. Increase of spontaneous intrachromosomal homologous recombination in mammalian cells expressing a mutant p53 protein. Oncogene 1997 ; 14 : 1117-22.
- 8. Mekeel KL, Tang W, Kachnic LA, et al. Inactivation of p53 results in high rates of homologous recombination. Oncogene 1997 ; 14 : 1847-57.
- 9. Gebow D, Miselis N, Liber HL. Homologous and nonhomologous recombination resulting in deletion : effects of p53 status, microhomology, and repetitive DNA length and orientation. Mol Cell Biol 2000 ; 20 : 4028-35.
- 10. Lu X, Lozano G, Donehower LA. Activities of wildtype and mutant p53 in suppression of homologous recombination as measured by a retroviral vector system. Mutat Res 2003 ; 522 : 69-83.
- 11. Saintigny Y, Rouillard D, Chaput B, et al. Mutant p53 proteins stimulate spontaneous and radiation-induced intrachromosomal homologous recombination independently of the alteration of the transactivation activity and of the G1 checkpoint. Oncogene 1999 ; 18 : 3553-63.
- 12. Lambert S, Lopez BS. Characterization of mammalian RAD51 double strand break repair using non lethal dominant negative forms. EMBO J 2000 ; 19 : 3090-9.
- 13. Wang YY, Maher VM, Liskay RM, McCormick JJ. Carcinogens can induce homologous recombination between duplicated chromosomal sequences in mouse L cells. Mol Cell Biol 1988 ; 8 : 196-202.
- 14. Akyuz N, Boehden GS, Susse S, et al. DNA substrate dependence of p53-mediated regulation of double-strand break repair. Mol Cell Biol 2002 ; 22 : 6306-17.
- 15. Michel B, Flores MJ, Viguera E, et al. Rescue of arrested replication forks by homologous recombination. Proc Natl Acad Sci USA 2001 ; 98 : 8181-8.
- 16. Saintigny Y, Delacote F, Vares G, et al. Characterization of homologous recombination induced by replication inhibition in mammalian cells. EMBO J 2001 ; 20 : 3861-70.
- 17. Saintigny Y, Lopez BS. Homologous recombination induced by replication inhibition is stimulated by expression of mutant p53. Oncogene 2002 ; 21 : 488-92.
- 18. Janz C, Wiesmuller L. Wild-type p53 inhibits replication-associated homologous recombination. Oncogene 2002 ; 21 : 5929-33.
- 19. Sengupta S, Linke SP, Pedeux R, et al. BLM helicase-dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J 2003 ; 22 : 1210-22.
- 20. Dudenhoffer C, Kurth M, Janus F, et al. Dissociation of the recombination control and the sequence-specific transactivation function of p53. Oncogene 1999 ; 18 : 5773-84.
- 21. Willers H, McCarthy EE, Wu B, et al. Dissociation of p53-mediated suppression of homologous recombination from G1/S cell cycle checkpoint control. Oncogene 2000 ; 19 : 632-9.
- 22. Linke SP, Sengupta S, Khabie N, et al. p53 interacts with hRAD51 and hRAD54, and directly modulates homologous recombination. Cancer Res 2003 ; 63 : 2596-605.
- 23. Yoon D, Wang Y, Stapleford K, et al. P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J Mol Biol 2004 ; 336 : 639-54.
- 24. Lee S, Cavallo L, Griffith J. Human p53 binds Holliday junctions strongly and facilitates their cleavage. J Biol Chem 1997 ; 272 : 7532-9.
- 25. Lee S, Elenbaas B, Levine A, Griffith J. p53 and its 14 kDa C-terminal domain recognize primary DNA damage in the form of insertion/deletion mismatches. Cell 1995 ; 81 : 1013-20.
- 26. Buchhop S, Gibson MK, Wang XW, et al. Interaction of p53 with the human Rad51 protein. Nucleic Acids Res 1997 ; 25 : 3868-74.
- 27. Bertrand P, Saintigny Y, Lopez BS. p53’s double life : transactivation-independent repression of homologous recombination. Trends Genet 2004 ; 20 : 235-43.
- 28. Dudenhoffer C, Rohaly G, Will K, et al. Specific mismatch recognition in heteroduplex intermediates by p53 suggests a role in fidelity control of homologous recombination. Mol Cell Biol 1998 ; 18 : 5332-42.
- 29. Susse S, Janz C, Janus F, et al. Role of heteroduplex joints in the functional interactions between human Rad51 and wild-type p53. Oncogene 2000 ; 19 : 4500-12.
- 30. Degtyareva N, Subramanian D, Griffith JD. Analysis of the binding of p53 to DNAs containing mismatched and bulged bases. J Biol Chem 2001 ; 276 : 8778-84.
- 31. Zink D, Mayr C, Janz C, Wiesmuller L. Association of p53 and MSH2 with recombinative repair complexes during S phase. Oncogene 2002 ; 21 : 4788-800.
- 32. Subramanian D, Griffith JD. Interactions between p53, hMSH2-hMSH6 and HMG I(Y) on Holliday junctions and bulged bases. Nucleic Acids Res 2002 ; 30 : 2427-34.
- 33. Waldman AS, Liskay RM. Dependence of intrachromosomal recombination in mammalian cells on uninterrupted homology. Mol Cell Biol 1988 ; 8 : 5350-7.
- 34. Rayssiguier C, Thaler DS, Radman M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch-repair mutants. Nature 1989 ; 342 : 396-401.
- 35. Jimenez GS, Nister M, Stommel JM, et al. A transactivation-deficient mouse model provides insights into Trp53 regulation and function. Nat Genet 2000 ; 26 : 37-43.
- 36. Komarov PG, Komarova EA, Kondratov RV, et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999 ; 285 : 1733-7.
- 37. Deng C, Zhang P, Harper JW, et al. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 1995 ; 82 : 675-84.
- 38. Szostak JW, Orr-Weaver TL, Rothstein RJ, Stahl FW. The double-strand-break repair model for recombination. Cell 1983 ; 33 : 25-35.
- 39. Rossignol JL. La recombinaison homologue : mécanismes et conséquences. Med Sci (Paris) 1990 ; 6 : 4-9.
List of figures
Figure 1
Modèle de réparation des cassures double brin (CDB) par recombinaison homomogue.
1. La CDB est métabolisée par une exonucléase qui produit des queues simple brin. 2. L’ADN simple brin envahit un ADN duplexe homologue intact. Cette étape requiert des séquences homologues, mais un polymorphisme limité est toléré par le processus : un échange de brin entre deux séquences non parfaitement homologues crée une molécule hybride porteuse de mésappariements de base, appelée hétéroduplexe, qui peut être prise en charge par le système de réparation des mésappariements de bases. Cette étape produit également des jonctions cruciformes (jonctions de Holliday) qui peuvent migrer (migration de branche), permettant ainsi l’élongation de la molécule hétéroduplexe. Cette étape initiale d’appariement, puis d’échange de brins, est catalysée par les protéines RecA chez la bactérie et Rad51 chez les eucaryotes. 3. Une polymérisation est déclenchée en utilisant l’extrémité 3’ du brin envahisseur comme amorce. Le brin d’ADN intact déplacé par la synthèse s’hybride alors avec la queue simple brin complémentaire, l’étape étant réalisée grâce à la protéine Rad54. Les cercles en pointillés indiquent les zones où la protéine p53 peut interagir directement avec l’intermédiaire de recombinaison homomogue. 4. Les brèches simple brin sont comblées par la polymérisation. 5. La conversion génique (transfert non réciproque d’information génétique ne modifiant pas la structure générale du chromosome) dépend de l’orientation de la réparation des mésappariements et de la séquence copiée dans la région de la CDB. Les jonctions de Holliday peuvent être résolues selon deux orientations alternatives (triangles noir ou blanc) : suivant l’orientation de résolution, les produits (conversion génique) seront associés on non à un crossing-over (échange réciproque d’information génétique) (d’après [38]).
Figure 2
Instabilité génétique associée à un excès de recombinaison homomogue.
A. Réarrangements chromosomiques résultant de crossing-over [39]. 1.Crossing-over inter-chromosomique ou entre chromatides soeurs décalées aboutissant à une amplification et à une délétion génique. 2.Crossing-over intrachromatidien entre séquence en répétition directe aboutissant à l’excision du fragment interne. 3.Crossing-over entre répétition en orientation inverse aboutissant à l’inversion du fragment interne. 4-5.Crossing-over interchromosomiques : selon l’orientation par rapport au centromère, les produits seront une translocation (4) ou un chromosome dicentrique et un chromosome acentrique (5). B. Conversion génique entre (1) deux hétéro-allèles aboutissant à une perte d’hétérozygotie ou entre (2) un pseudogène (flèche jaune) et un gène (flèche bleue), aboutissant à l’inactivation du gène. Les mutations transférées sont représentées par le trait rouge.
Figure 3
Structure de p53 et des protéines de recombinaison homologue Rad51 (humaine) et RecA (bactérienne).
A. Domaines fonctionnels de la protéine p53. SLN : signal de localisation nucléaire. B. Domaines fonctionnels de Rad51 et de RecA. Le domaine central, conservé au cours de l’évolution, est appelé « boucle RecA ». Les nombres indiquent la position des résidus d’acides aminés.
Figure 4
Complexes protéiques de recombinaison homomogue chez les mammifères.
A. Les cinq paralogues (Xrcc2, Xrcc3, Rad51B, Rad51C et Rad51D) de la protéine Rad51 sont impliqués dans deux complexes différents. Aucune interaction physique entre ces paralogues et la protéine p53 n’a clairement été démontrée. B. La protéine Rad51 interagit physiquement avec les autres protéines contrôlant la recombinaison homomogue. Les protéines du complexe capables d’interagir physiquement avec p53 sont cerclées de rouge.